Introduction
Machado-Joseph Disease (MJD), also known as Spinocerebellar Ataxia Type 3, is a genetic disorder caused by a mutation in the ATXN3 gene. It leads to neurodegeneration, primarily affecting the cerebellum and brainstem. Symptoms vary with different types of MJD and can include ataxia, spasticity, neuropathy, and sleep disorders. Diagnosis involves clinical evaluation, neuroimaging, and genetic testing. Management focuses on supportive care, including medications, therapies, and assistive devices. Ongoing research aims to understand the genetic mechanisms underlying MJD and develop targeted treatments.
Machado-Joseph Disease
Formal Name: Spinocerebellar Ataxia Type 3 (SCA3)
Demographic Information
- Incidence: Estimates vary, but it is considered one of the most common autosomal dominant ataxias worldwide.
- Prevalence: Particularly prevalent in people of Portuguese or Azorean descent. For example, on the island of Flores in the Azores, the prevalence is about 1 in 140 people.
- Gender: Affects both males and females equally.
- Onset Age: Symptoms can begin at any age, but typically start between the ages of 10 and 70, depending on the type.
Coding
- ICD-11: 8A44.0
- ICD-10-CM: G11.2
- OMIM: 109150 - Machado-Joseph Disease
- UMLS: C0752166
- MeSH: D020611
- GARD: 7911
Medical Features and Pathophysiology
Etiology: Machado-Joseph Disease (MJD) is caused by an expansion of the CAG trinucleotide repeat in the ATXN3 gene. This mutation leads to the production of an abnormal version of the ataxin-3 protein, which aggregates in neurons and causes neurodegeneration. The disease is inherited in an autosomal dominant pattern, meaning one copy of the altered gene is sufficient to cause the disorder.
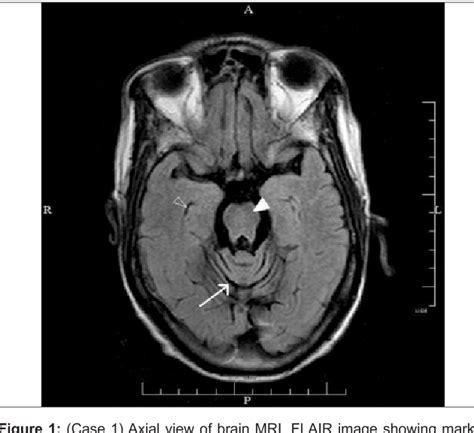
Pathology: The degeneration primarily affects the cerebellum and brainstem, leading to progressive ataxia, spasticity, and other neurological symptoms. The accumulation of abnormal protein clumps in neurons disrupts normal cellular function and ultimately leads to cell death.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Genetic
- Pathology: Heritable
- Rarity Classification: Rare
- Typical Onset: Adulthood
- Gender Impact: Mostly Men
- Progressive/Degenerative: Yes
- Seizure Prevalence: No (<10%)
- Population Trend: Unknown
Pathophysiology
SCA3 gene mutation in the Azores, and is found in other places that suggest origination in Portugal's Sephardic Jewish community. ATXN3 also implicated.
AAC Considerations
Recommended Access Modalities: All - Variable
Additional Clinical Notes
There are five subtypes of presentation, ranging from fatal <10yrs (13%) to Parkisonian type presentation. 57% have type II, which is what I primarily address here. Uniquely likely among those of Portuguese descent, particularly in the Azores
Symptoms
Type I (MJD-I)
- Onset Age: 10-30 years
- Characteristics: Rapidly progressive with severe dystonia, spasticity, and early disability.
Type II (MJD-II)
- Onset Age: 20-50 years
- Characteristics: Gradually progressive with spasticity, ataxia, and difficulty walking.
Type III (MJD-III)
- Onset Age: 40-70 years
- Characteristics: Slowly progressive with neuropathy, muscle atrophy, and less severe ataxia.
Common Symptoms
- Gait and limb ataxia
- Dysarthria (difficulty speaking)
- Nystagmus and other eye movement abnormalities
- Spasticity and rigidity
- Neuropathy causing numbness, tingling, and pain
- Sleep disorders and dysphagia (difficulty swallowing)
- Autonomic dysfunction affecting bladder control and heart rate
Diagnosis
- Clinical Evaluation: Based on symptom presentation, family history, and neurological examination.
- Neuroimaging: MRI and CT scans can reveal cerebellar and brainstem atrophy.
- Genetic Testing: Confirms the diagnosis by identifying the CAG repeat expansion in the ATXN3 gene. Genetic counseling is recommended for affected individuals and their families to discuss the implications of the test results.
Management and Treatment
Supportive Care: Focuses on managing symptoms and maintaining quality of life.
- Medications: Used to control symptoms such as spasticity, pain, and sleep disturbances.
- Physical Therapy: Helps maintain mobility and balance.
- Occupational Therapy: Assists with daily living activities and the use of adaptive devices.
- Speech Therapy: Addresses dysarthria and swallowing difficulties.
- Assistive Devices: Wheelchairs, walkers, and other mobility aids.
Assistive Technology Suggestions
- Communication Devices: AAC devices for those with severe speech impairments.
- Environmental Control Systems: Adaptive switches and voice-activated systems to enhance independence.
- Mobility Aids: Wheelchairs, walkers, and canes to assist with mobility and prevent falls.
Access Modalities
Touchscreen devices work well in early stages despite mild ataxia. As the condition progresses, switch access or adapted keyboards may be needed to compensate for tremor and spasticity. Voice-controlled technology can be useful if dysarthria remains mild, though speech typically degrades over time. Access needs should be reassessed regularly as motor function declines.
Comprehensive Management and Care Strategies
Medical Management: - Monitoring disease progression and emerging complications - Pain management for neuropathy and spasticity - Fall prevention and mobility support
Behavioral and Psychological Support: - Counseling for progressive loss of function and genetic implications - Support groups for individuals and families - Genetic counseling for family planning decisions
Challenges and Considerations
The progressive nature of MJD requires proactive planning for mobility loss, swallowing difficulties, and communication challenges. Genetic inheritance patterns necessitate family counseling. The variability in age of onset and disease severity complicates prognostic discussions. Individuals often face social isolation as physical limitations increase.
Future Directions and Research
Research focuses on gene silencing therapies to reduce toxic protein aggregation, neuroprotective agents to slow degeneration, and biomarkers for earlier diagnosis. Clinical trials are exploring antisense oligonucleotides and small molecule interventions targeting the ATXN3 mutation.
References
- National Organization for Rare Disorders (NORD): Provides an overview of Machado-Joseph Disease, including symptoms, diagnosis, and treatment options. NORD MJD
- National Institute of Neurological Disorders and Stroke (NINDS): Offers insights into the causes and treatment of Machado-Joseph Disease. NINDS MJD
- Verywell Health: Discusses symptoms, diagnosis, and management of Machado-Joseph Disease. Verywell Health MJD
- National Ataxia Foundation: Provides resources and information on spinocerebellar ataxia type 3 (SCA3). National Ataxia Foundation
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Machado-Joseph Disease?
What does Machado-Joseph Disease do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.