Introduction
MECP2 Duplication Syndrome, affecting primarily males at an estimated 1 in 150,000 live births, is caused by a duplication of the MECP2 gene on the X chromosome, leading to overproduction of the MeCP2 protein and disrupting normal neurological and developmental functions. Symptoms range from intellectual disability, developmental delays, seizures, and behavioral issues to recurrent respiratory infections and feeding difficulties. Diagnosis involves clinical evaluation, genetic testing, and neuroimaging. Management focuses on supportive care, including anticonvulsants, therapy, respiratory and nutritional support, and assistive technology. Future research is focused on understanding the genetic mechanisms and exploring new treatments, including gene therapy and neuroprotective strategies.
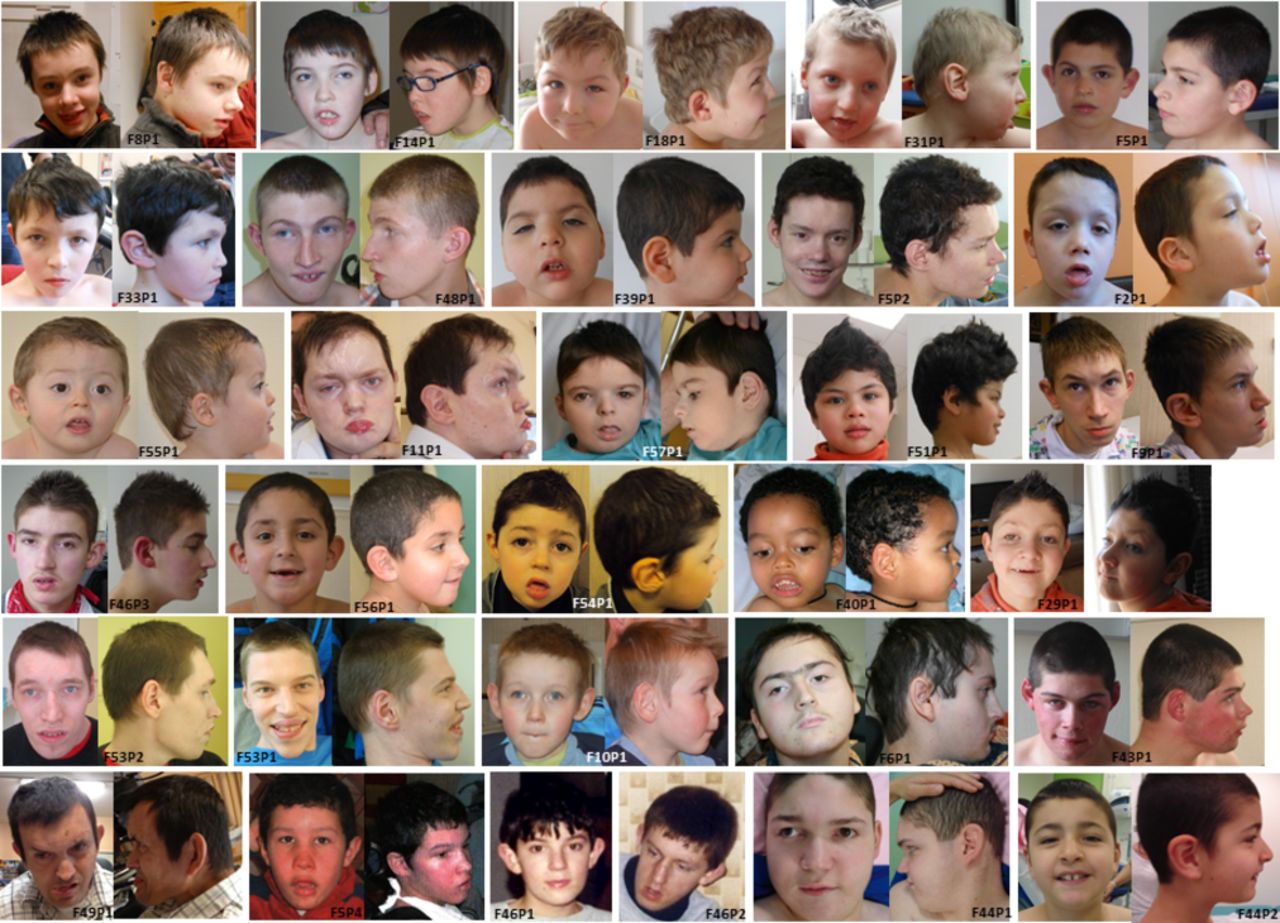
MECP2 Duplication Syndrome
Demographic Information
- Incidence: Estimated to affect 1 in 150,000 live births, primarily in males due to the X-linked pattern of inheritance.
- Prevalence: Approximately 120 known cases worldwide, though this may be an underestimate due to underdiagnosis.
- Gender: Predominantly affects males; females can be carriers and may exhibit milder symptoms due to X-inactivation.
- Onset Age: Symptoms can be present from birth or early infancy, typically diagnosed in early childhood.
Coding
- ICD-11: LD40.5
- ICD-10-CM: Q99.8
- OMIM: 300260 - MECP2 Duplication Syndrome
- UMLS: C3280328
- MeSH: D000926
- GARD: 9382
Medical Features and Pathophysiology
- Etiology: MECP2 Duplication Syndrome is caused by a duplication of the MECP2 gene on the X chromosome (Xq28). This duplication leads to an overproduction of the MeCP2 protein, disrupting normal neurological and developmental functions. The syndrome is typically inherited in an X-linked manner, although de novo duplications can also occur.
- Pathology: The overexpression of the MeCP2 protein interferes with normal brain development and function, leading to a wide range of neurological symptoms. This includes the impairment of synaptic function and neuronal communication, resulting in cognitive and motor deficits.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Genetic
- Pathology: Heritable
- Rarity Classification: Ultra-Orphan
- Typical Onset: Birth
- Gender Impact: Mostly Men
- Seizure Prevalence: Yes (>90%)
- Population Trend: Unknown
Pathophysiology
MECP2
AAC Considerations
Recommended Access Modalities: All - Variable
Additional Clinical Notes
Women with this mutation generally unaffected
Patient Advocacy & Support Organizations
- http://mecp2duplicationuk.org.uk/
- https://www.rarechromo.org/
- https://www.facebook.com/groups/mecp2families/
- https://www.facebook.com/groups/509218079250240/
Symptoms
Neurological Symptoms:
- Intellectual disability, ranging from moderate to severe.
- Developmental delays, particularly in motor and speech skills.
- Hypotonia (low muscle tone) in infancy, progressing to spasticity in childhood.
- Seizures, which can be refractory and vary in type (generalized tonic-clonic, atonic, absence, and myoclonic seizures).
- Autistic features and stereotypic movements, including midline hand movements.
- Behavioral and psychiatric symptoms such as mood disorders and abnormal movements (e.g., choreiform movements).
Other Symptoms:
- Recurrent respiratory infections, often leading to pneumonia and necessitating hospitalization.
- Feeding difficulties, which may require nutritional support via gastrostomy.
- Dysphagia (difficulty swallowing).
- Gastroesophageal reflux disease (GERD).
- Progressive spasticity, particularly in the lower limbs, leading to mobility issues and the potential need for a wheelchair.
Diagnosis
- Clinical Evaluation: Based on a combination of neurological and developmental symptoms.
- Genetic Testing: Confirms the presence of the MECP2 duplication on the X chromosome. This is typically done through array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA).
- Neuroimaging: MRI and CT scans may show abnormalities such as cortical atrophy, ventricular dilatation, and cerebellar atrophy.
Management and Treatment
- Supportive Care: Focuses on managing symptoms and improving quality of life.
- Anticonvulsants: To manage seizures, although they can be refractory to treatment.
- Physical and Occupational Therapy: To maintain mobility and daily functioning.
- Speech Therapy: To address communication difficulties and improve feeding.
- Respiratory Support: For recurrent infections, including potential use of antibiotics and respiratory therapies.
- Nutritional Support: May require gastrostomy feeding for severe cases of dysphagia and GERD.
- Experimental Treatments: Gene therapy and other molecular therapies are being investigated, aiming to correct the overexpression of the MeCP2 protein.
Assistive Technology Suggestions
- Communication Devices: AAC devices for those with severe speech impairments.
- Environmental Control Systems: Adaptive switches and voice-activated systems to enhance independence in daily activities.
- Mobility Aids: Wheelchairs and walkers to assist with mobility and prevent falls.
Access Modalities
Motor control varies significantly, requiring individualized assessment. Many benefit from touchscreen devices with simplified interfaces. Switch access may be necessary for those with severe spasticity or limited range of motion. Voice control is generally not viable given the prevalence of severe speech impairment. Access methods should anticipate progressive motor decline.
Comprehensive Management and Care Strategies
Medical Management: - Routine monitoring of seizures, infections, and spasticity - Respiratory infection prevention and prompt treatment - Nutritional support for feeding difficulties and reflux
Family Support: - Genetic counseling for X-linked inheritance pattern - Support groups for families navigating complex medical needs - Respite care resources
Challenges and Considerations
Recurrent respiratory infections drive significant morbidity and hospitalization. Seizures are often refractory to standard treatments. Progressive spasticity limits mobility over time. The X-linked pattern means carrier mothers face reproductive counseling decisions. Educational programming must account for severe intellectual disability and limited communication capacity.
Future Directions and Research
Research explores gene therapy to normalize MeCP2 protein levels, antisense oligonucleotides to reduce overexpression, and strategies to mitigate immune dysfunction. Clinical trials are investigating treatments to reduce seizure burden and prevent respiratory complications.
References
- National Organization for Rare Disorders (NORD): Provides an overview of MECP2 Duplication Syndrome, including symptoms, diagnosis, and treatment options.
- MedlinePlus Genetics: Offers detailed information on the genetic aspects and clinical features of MECP2 Duplication Syndrome.
- Children's Hospital Colorado: Discusses the clinical management and supportive care strategies for children with MECP2 Duplication Syndrome.
- Karger Publishers: Provides insights into the neurological symptoms, neuroradiology, and recurrent infections associated with MECP2 Duplication Syndrome.
Epidemiology and Demographics
Etiology and Pathophysiology
What causes MECP2 Duplication Syndrome?
What does MECP2 Duplication Syndrome do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.