Introduction
Mowat-Wilson Syndrome (MWS) is a rare genetic disorder affecting 1 in 50,000 to 70,000 live births. It's caused by mutations in the ZEB2 gene and is characterized by distinctive facial features, intellectual disability, and various congenital anomalies. Symptoms are typically apparent at birth or in early childhood and include severe intellectual disability, delayed motor skills, seizures, and various physical abnormalities. Diagnosis involves clinical evaluation, genetic testing, and neuroimaging. While there's no cure, management focuses on symptom control and improving quality of life through surgery, medication, therapy, and assistive technology. Ongoing research aims to better understand the genetic mechanisms of MWS and develop targeted treatments.
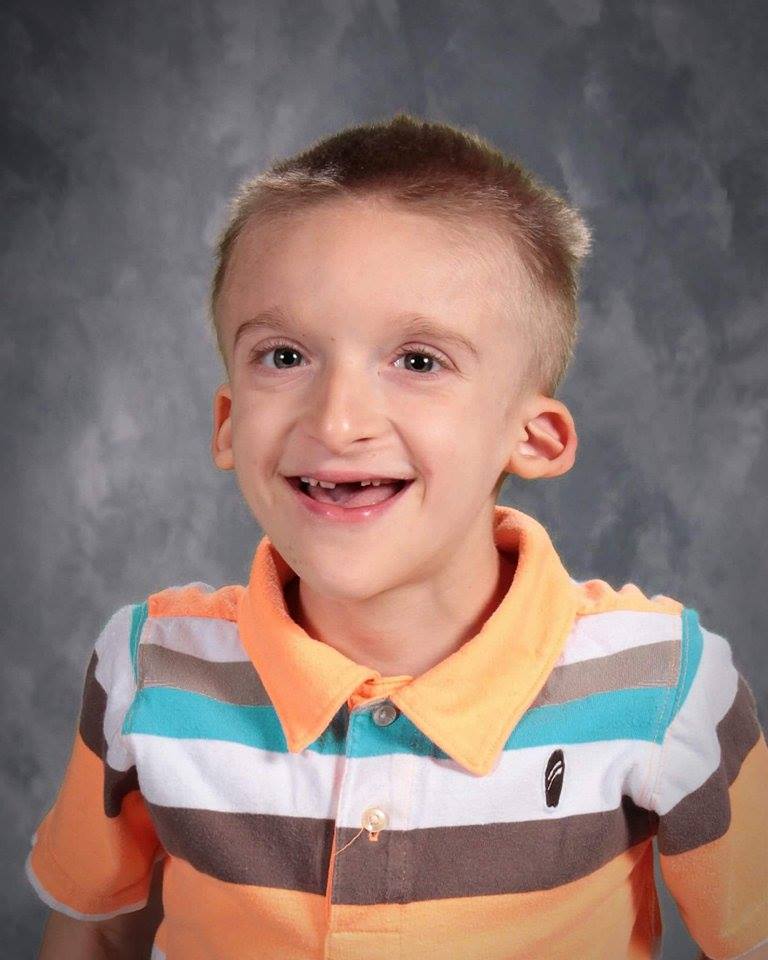
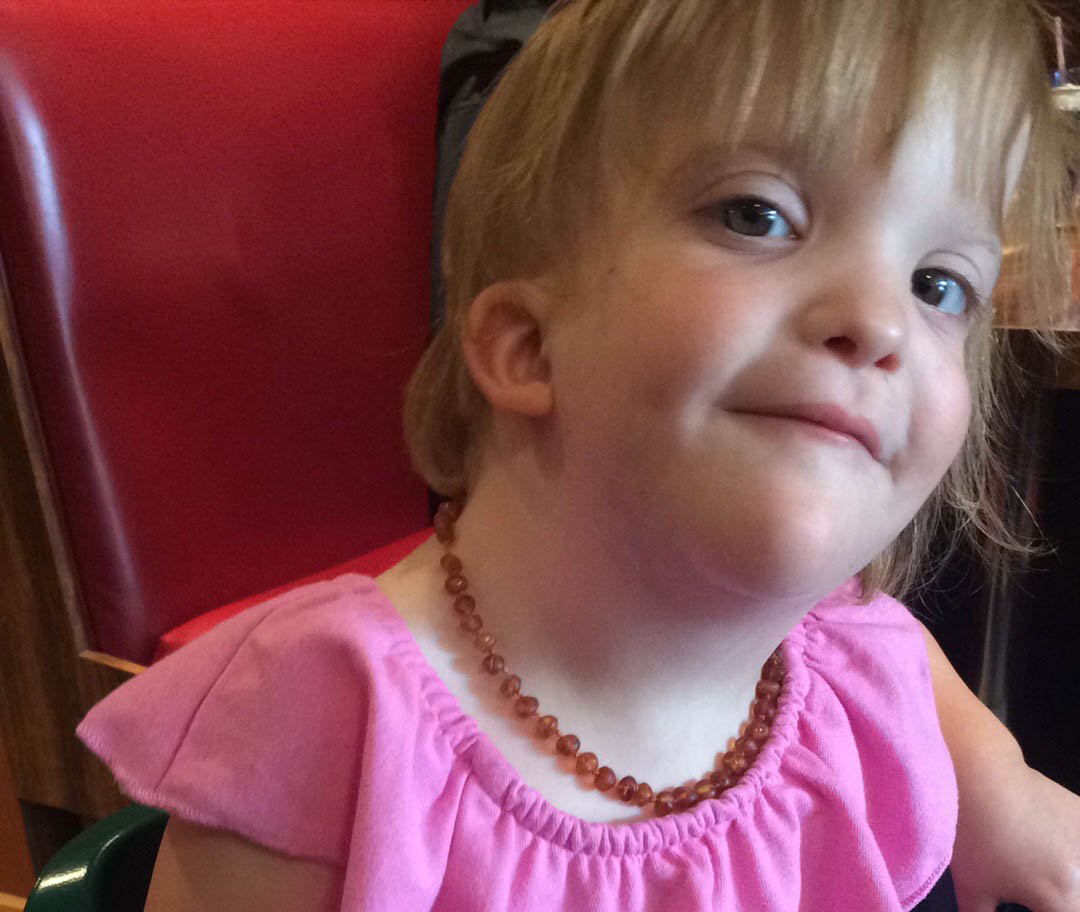
Mowat-Wilson Syndrome
Demographic Information
- Incidence: Approximately 1 in 50,000 to 1 in 70,000 live births.
- Prevalence: Several hundred cases reported worldwide, though this may be an underestimate due to underdiagnosis.
- Gender: Affects both males and females equally.
- Onset Age: Symptoms are typically apparent at birth or in early childhood.
Coding
- ICD-11: LD50.1
- ICD-10-CM: Q87.89
- OMIM: 235730 - Mowat-Wilson Syndrome
- UMLS: C536990
- MeSH: D057801
- GARD: 6249
Medical Features and Pathophysiology
Etiology
Mowat-Wilson Syndrome is caused by mutations or deletions in the ZEB2 gene located on chromosome 2q22.3. This gene encodes a transcription factor critical for various developmental processes. Most cases are de novo mutations, meaning they occur spontaneously and are not inherited from parents.
Pathology
The genetic mutations result in disrupted development of multiple organ systems. The syndrome is characterized by distinctive facial features, intellectual disability, and various congenital anomalies, including Hirschsprung disease, congenital heart defects, and genitourinary abnormalities.
Symptoms
Facial Features
- High forehead, wide-spaced deep-set eyes, broad nasal bridge with a rounded tip, prominent chin, large and uplifted earlobes with a dimple in the middle.
- Facial features become more distinctive with age, and affected individuals often have a smiling, open-mouthed expression.
Neurological Symptoms
- Intellectual disability, typically severe, though some may have moderate intellectual disability.
- Delayed development of motor skills, such as sitting, standing, and walking.
- Absent or severely limited speech; some may learn to speak a few words, while others may use sign language or other forms of communication.
- Seizures, which may be variable in type and can include absence, generalized, and myoclonic seizures. Seizures are often resistant to treatment in childhood but may be better managed in adulthood.
Other Symptoms
- Hirschsprung disease (present in about 50% of individuals), causing severe constipation and intestinal blockage.
- Congenital heart defects, including patent ductus arteriosus and ventricular septal defects.
- Genitourinary anomalies such as hypospadias and cryptorchidism in males.
- Microcephaly (small head size), short stature, and chronic constipation.
- Structural brain anomalies, including agenesis of the corpus callosum, ventriculomegaly, and cortical atrophy.
Diagnosis
- Clinical Evaluation: Diagnosis is based on clinical presentation, including characteristic facial features and congenital anomalies.
- Genetic Testing: Confirms the diagnosis by identifying mutations in the ZEB2 gene. Methods include single gene testing, multi-gene panels, chromosomal microarray analysis (CMA), and exome sequencing.
- Neuroimaging: MRI and CT scans to identify brain abnormalities. Cardiac and renal ultrasounds to detect associated anomalies.
Management and Treatment
Supportive Care
Focuses on managing symptoms and improving quality of life. There is no cure for MWS.
- Surgery: For congenital heart defects, Hirschsprung disease, and other anatomical anomalies.
- Medications: To control seizures and manage other symptoms.
- Physical and Occupational Therapy: To improve motor skills and daily functioning.
- Speech Therapy: To assist with communication difficulties.
- Nutritional Support: For feeding difficulties and chronic constipation, including possible gastrostomy.
Assistive Technology Suggestions
- Communication Devices: Augmentative and Alternative Communication (AAC) devices for those with severe speech impairments.
- Mobility Aids: Wheelchairs and walkers to assist with mobility.
- Environmental Control Systems: Adaptive switches and voice-activated systems to enhance independence.
Access Modalities
Most individuals have significant intellectual disability affecting literacy and symbol recognition. Simple cause-and-effect switch activities may be appropriate for some. Eye gaze can work for individuals with better motor control but requires careful cognitive assessment. Touchscreen devices with highly simplified interfaces using photos or single-meaning symbols are often most successful. Partner-assisted scanning may be needed for those with severe motor and cognitive limitations.
Comprehensive Management and Care Strategies
Medical Management: - Monitoring cardiac and renal function - Management of Hirschsprung disease and chronic constipation - Seizure control and developmental support
Family Support: - Genetic counseling regarding recurrence risk - Connection to syndrome-specific support networks - Coordination of surgical and medical interventions
Challenges and Considerations
Hirschsprung disease requires surgical intervention and ongoing bowel management. Seizures can be difficult to control. Severe intellectual disability limits communication and self-care abilities. Congenital anomalies often require multiple surgeries in infancy and childhood. Educational programming must be highly individualized with significant modification and support.
Future Directions and Research
Research explores the role of ZEB2 in development, potential gene therapy approaches, and improved seizure management strategies. Natural history studies are documenting the full spectrum of clinical features and outcomes to guide medical management.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Genetic
- Pathology: Heritable
- Rarity Classification: Ultra-Orphan
- Typical Onset: Birth
- Gender Impact: Either Gender
- Seizure Prevalence: Yes (>90%)
- Population Trend: Unknown
Pathophysiology
ZEB2 gene deletion, which is associated with failure to transcribe proteins critical to organ and tissue development. Autosomal dominant.
AAC Considerations
Recommended Access Modalities: Switch,Eye Gaze
Additional Clinical Notes
ZEB2 gene mutation; major motor, intellectual, and intestinal complications
Patient Advocacy & Support Organizations
References
- National Organization for Rare Disorders (NORD): Provides an overview of Mowat-Wilson Syndrome, including symptoms, diagnosis, and treatment options. NORD MWS
- Genetic and Rare Diseases Information Center (GARD): Detailed information on the genetic aspects and clinical features of Mowat-Wilson Syndrome. GARD MWS
- MedlinePlus Genetics: Discusses the clinical management and supportive care strategies for Mowat-Wilson Syndrome. MedlinePlus MWS
- Mowat-Wilson Syndrome Foundation: Offers resources, support, and information for families and caregivers. MWS Foundation
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Mowat-Wilson syndrome?
What does Mowat-Wilson syndrome do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.