Introduction
Multiple System Atrophy (MSA) is a sporadic, progressive neurodegenerative disorder affecting multiple parts of the nervous system. It's characterized by the accumulation of abnormal alpha-synuclein protein in glial cells, leading to cell damage and death. Clinical features include autonomic dysfunction, motor abnormalities resembling Parkinson's disease, and cerebellar ataxia. Diagnosis relies on clinical evaluation and characteristic symptom patterns, with neuroimaging providing supportive evidence. Most individuals with MSA eventually require assistive technology as motor and autonomic impairments progress.
Multiple System Atrophy (MSA)
Demographic Information
- Incidence: Approximately 3-4 cases per 100,000 people per year
- Prevalence: Estimated 4-5 cases per 100,000 people
- Gender: Slightly more common in males (male-to-female ratio approximately 1.4:1)
- Onset Age: Typically begins in the late 50s to early 60s
Coding
- ICD-11: 8A06.2
- ICD-10-CM: G90.3
- OMIM: 146500
- UMLS: C0751964
- MeSH: D020528
- GARD: 7198
Medical Features and Pathophysiology
Etiology
Multiple System Atrophy (MSA) is a sporadic, progressive neurodegenerative disorder with no known familial inheritance. The exact cause of MSA is unknown, but it is characterized by the accumulation of abnormal alpha-synuclein protein in glial cells, leading to cell damage and death. Environmental factors and genetic predispositions may contribute, although no specific cause has been definitively identified.
Pathology
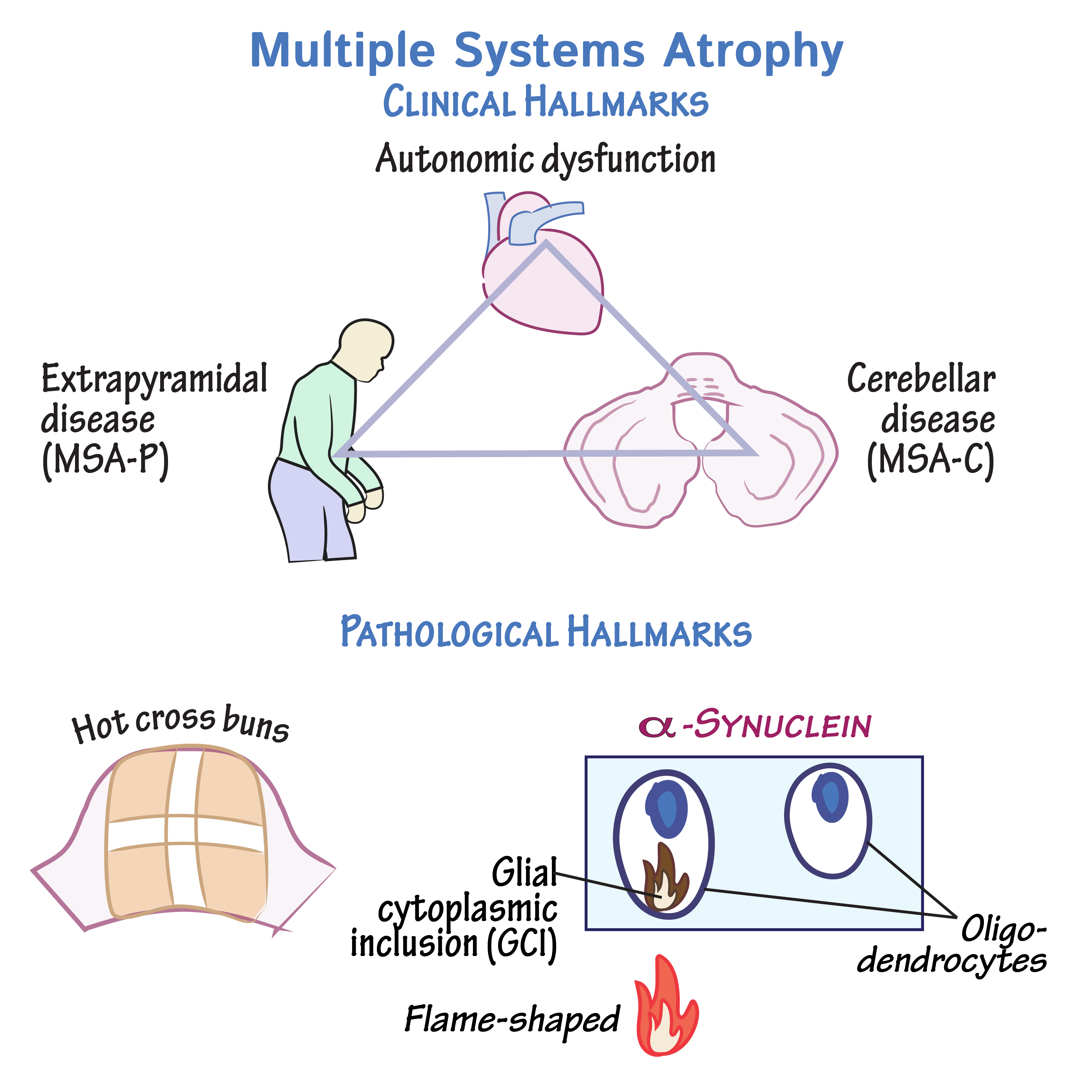
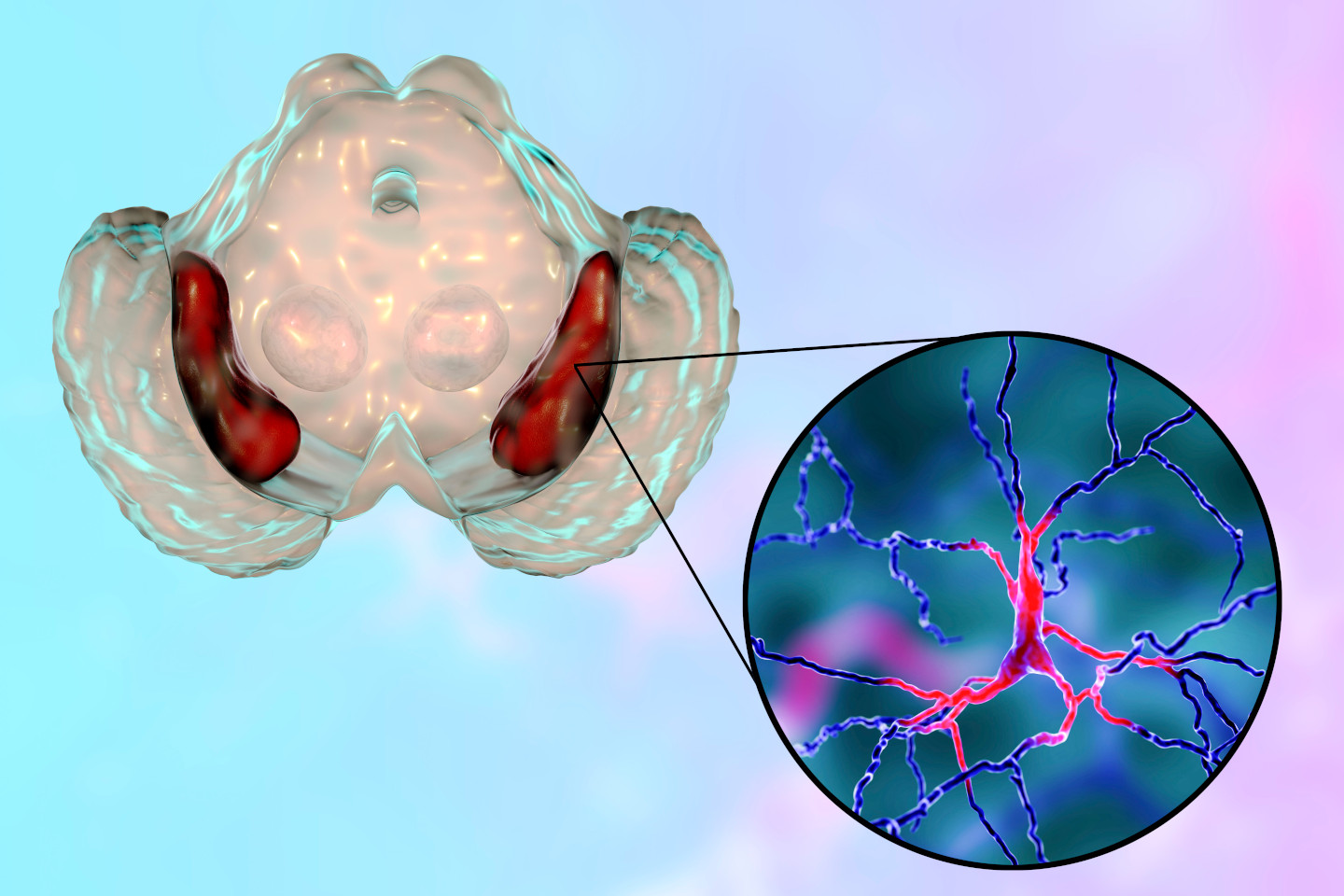
MSA affects multiple parts of the nervous system, including the autonomic nervous system, basal ganglia, cerebellum, and brainstem. The pathological hallmark of MSA is the presence of glial cytoplasmic inclusions (GCIs) composed of alpha-synuclein. These inclusions disrupt normal cellular functions, leading to widespread neurodegeneration. The specific areas affected dictate the clinical presentation and subtype of MSA: MSA-P (parkinsonian) and MSA-C (cerebellar).
Symptoms
- Early Symptoms:
- Autonomic dysfunction (e.g., orthostatic hypotension, urinary incontinence)
- Motor abnormalities similar to Parkinson's disease (e.g., bradykinesia, rigidity, tremor)
- Cerebellar ataxia (e.g., unsteady gait, coordination difficulties)
- Progressive Symptoms:
- Severe autonomic failure (e.g., profound blood pressure fluctuations, severe bladder dysfunction)
- Progressive motor impairments (e.g., increasing rigidity, bradykinesia, postural instability)
- Speech and swallowing difficulties (dysarthria, dysphagia)
- Respiratory complications, including stridor and sleep apnea
Diagnosis
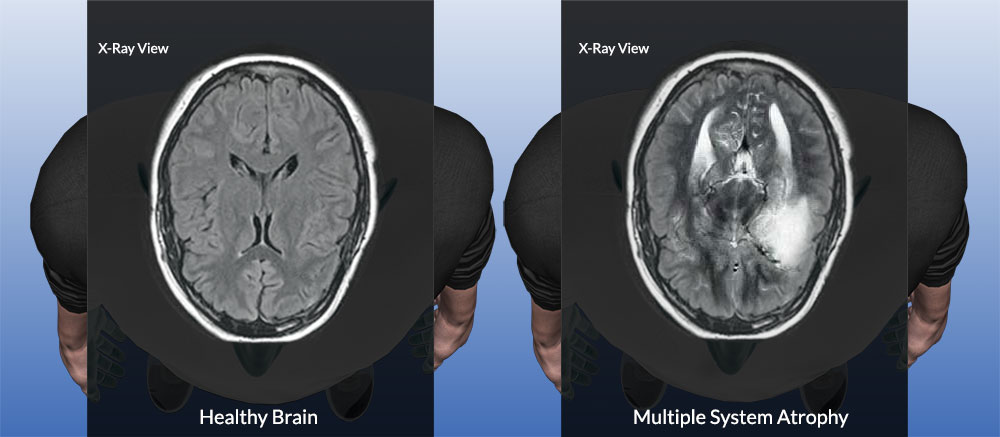
MSA is diagnosed based on clinical evaluation and the presence of characteristic symptoms. There is no specific test for MSA, but MRI can show atrophy in specific brain regions, such as the cerebellum and putamen. Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT) can assess functional changes in the brain. Autonomic testing may also be used to evaluate the extent of autonomic dysfunction. A comprehensive neurological examination is essential for differential diagnosis to exclude other conditions such as Parkinson's disease and pure autonomic failure.
Assistive Suggestions and Requirements
Requirement Percentage for Assistive Technology
A significant percentage of patients with MSA will eventually require some form of assistive technology due to progressive motor and autonomic impairments.
Assistive Technology Suggestions
- Mobility Aids:
- Rollators or Walkers: To aid with balance and prevent falls.
- Wheelchairs: For advanced stages when walking becomes difficult.
- Communication Aids:
- Voice Amplifiers: To assist with soft or unclear speech.
- Speech Generating Devices (SGDs): For those who lose the ability to speak clearly.
- Home Modifications:
- Grab Bars and Handrails: To enhance safety, especially in bathrooms.
- Adjustable Beds and Reclining Chairs: To improve comfort and ease of movement.
- Feeding Aids:
- Adaptive Utensils and Plates: For individuals with reduced dexterity and coordination.
Access Modalities
- Switch Access: Useful for individuals with severe motor impairments to control communication devices and computers.
- Eye-Tracking Systems: Beneficial for those who retain eye movement control but have lost most other motor functions.
- Touchscreen Devices: May be useful in the earlier stages when fine motor skills are still relatively preserved.
Care Management and Therapeutic Techniques
Aims
- To manage symptoms and maintain the highest possible level of independence.
- To provide supportive care and improve quality of life.
- To offer education and support to patients and caregivers.
SLP Suggestions
- Assessment and Intervention:
- Regular Speech and Swallowing Assessments: To monitor changes and adjust therapy plans.
- Swallowing Techniques: To ensure safe swallowing and reduce aspiration risk.
- Voice Therapy: Exercises to maintain voice strength and clarity.
- Augmentative and Alternative Communication (AAC):
- Introduction of AAC devices early in the disease progression for familiarity and ease of use.
Special Educator Suggestions
- Cognitive Rehabilitation:
- Activities to enhance executive function, memory, and attention.
- Use of memory aids like notebooks or electronic organizers.
- Behavioral Strategies:
- Structured routines to manage apathy and maintain engagement.
- Positive reinforcement to encourage participation and effort.
Occupational Therapist Suggestions
- Daily Living Skills:
- Training in adaptive equipment use for self-care activities (e.g., dressing, grooming).
- Techniques to conserve energy and manage fatigue.
- Home and Environmental Modifications:
- Assessing and modifying the home for safety and accessibility.
- Recommendations for ergonomic furniture and tools to support independence.
Recommendations on AAC and Other Details
- Text-Based AAC:
- Use of text-to-speech apps and devices for individuals with good literacy skills.
- Predictive text features to speed up communication.
- Symbol-Based AAC:
- For individuals with cognitive impairments affecting literacy, symbol-based systems like Picture Communication Symbols (PCS) can be useful.
- Dynamic display devices that can grow with the user’s needs.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Ideopathic
- Pathology: Structural
- Rarity Classification: Rare
- Typical Onset: Adulthood
- Gender Impact: Either Gender
- Progressive/Degenerative: Yes
- Seizure Prevalence: No (<10%)
- Population Trend: Unknown
Pathophysiology
CNS damage possibly secondary to overexpression of alpha-synuclein protein
AAC Considerations
Recommended Access Modalities: All - Progression
Additional Clinical Notes
Also called Shy-Drager syndrome; presents similar to other Parkinsonian and cerebellar neurodegenerative disorders
References
- , & . (2016). Review: Multiple system atrophy: emerging targets for interventional therapies. Neuropathology and Applied Neurobiology. https://doi.org/10.1111/nan.12304
- , et al. (2006). HFE H63D polymorphism is increased in patients with amyotrophic lateral sclerosis of Italian origin. Journal of Neurology Neurosurgery & Psychiatry. https://doi.org/10.1136/jnnp.2006.092338
- fanciulli. (2015).
- https://www.ncbi.nlm.nih.gov/pubmed/23192520 https://rarediseases.org/rare-diseases/multiple-system-atrophy/
Additional Information
MSA requires coordinated multidisciplinary care. Regular neurological follow-up combined with speech-language pathology, occupational therapy, and physical therapy can help manage symptoms and maintain function as the disease progresses. Patient and caregiver education through support groups and community resources provides practical and emotional support.
Extended Information
Pathological Insights and Disease Mechanism
MSA is characterized by the accumulation of alpha-synuclein in oligodendrocytes, forming glial cytoplasmic inclusions (GCIs). These inclusions interfere with the normal functioning of glial cells and neurons, leading to widespread neurodegeneration. The disease affects various regions of the brain, including the basal ganglia, cerebellum, and brainstem, resulting in a range of motor and autonomic symptoms. The exact mechanisms leading to the abnormal accumulation of alpha-synuclein and subsequent neurodegeneration are not fully understood, but ongoing research is focused on uncovering these processes.
Genetic and Environmental Factors
While MSA is generally considered sporadic, some genetic predispositions have been identified. Research has shown that certain polymorphisms in genes related to alpha-synuclein processing and clearance may increase the risk of developing MSA. Environmental factors, such as exposure to neurotoxins, have also been implicated, although their role is less clear. Understanding the interplay between genetic and environmental factors is crucial for developing preventive and therapeutic strategies.
Clinical Presentation and Disease Progression
The clinical presentation of MSA varies depending on the predominant symptoms, leading to the classification into two main subtypes: MSA-P (parkinsonian) and MSA-C (cerebellar). MSA-P is characterized by symptoms similar to Parkinson's disease, including bradykinesia, rigidity, and tremor. MSA-C is marked by cerebellar ataxia, manifesting as unsteady gait, poor coordination, and speech difficulties. Autonomic dysfunction is a hallmark of both subtypes and can include orthostatic hypotension, urinary incontinence, and erectile dysfunction. As the disease progresses, symptoms worsen, and patients may experience severe motor impairments, profound autonomic failure, and respiratory complications.
Differential Diagnosis and Overlapping Syndromes
MSA shares clinical features with Parkinson's disease, pure autonomic failure, and other cerebellar ataxias, making diagnosis challenging. Detailed history and neurological examination are essential. MRI findings such as putaminal atrophy and cerebellar shrinkage support the diagnosis but are not always present in early stages.
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Multiple System Atrophy?
What does Multiple System Atrophy do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.