Introduction
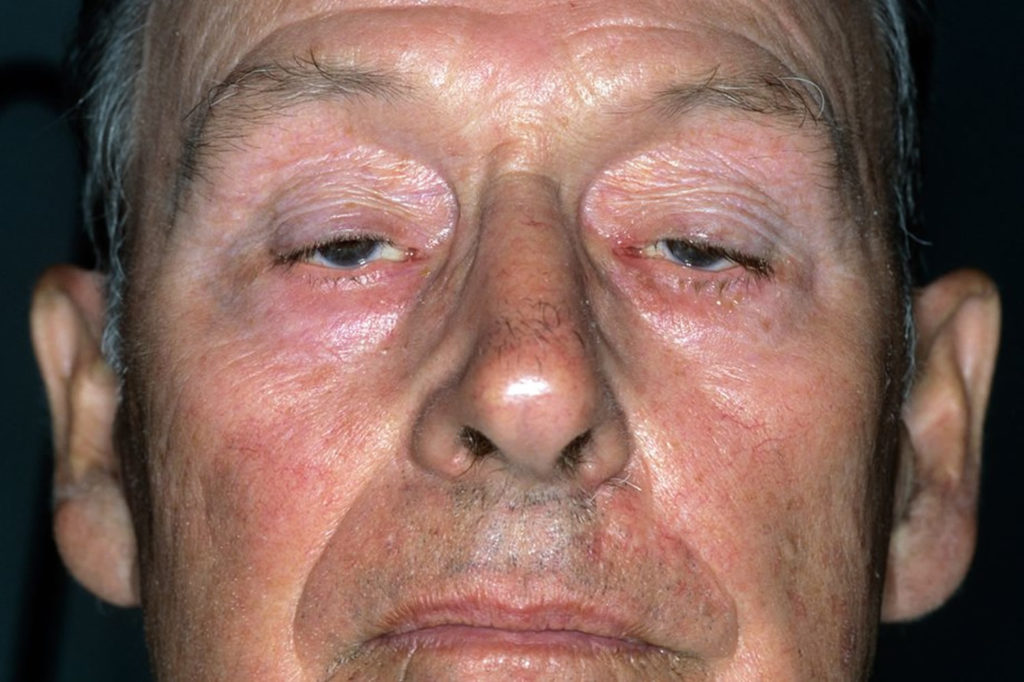
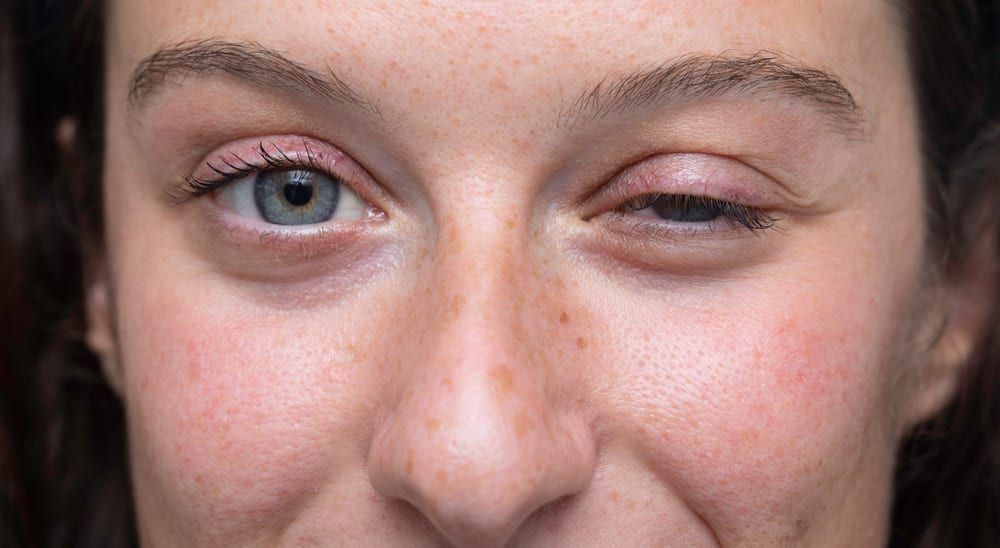
Myasthenia Gravis (MG) is an autoimmune disorder characterized by fluctuating muscle weakness and fatigue. Antibodies target acetylcholine receptors at the neuromuscular junction, impairing signal transmission from nerves to muscles. The condition shows a bimodal age distribution, affecting women more commonly in their 20s-30s and men in their 60s-70s. Symptoms typically begin with ocular weakness (ptosis, diplopia) and may progress to involve bulbar, limb, and respiratory muscles. Diagnosis relies on antibody testing, electrophysiological studies, and clinical response to acetylcholinesterase inhibitors.
Myasthenia Gravis
Demographic Information
- Incidence: Approximately 1 in 10,000 to 1 in 20,000 people per year
- Prevalence: About 20 cases per 100,000 people
- Gender: More common in women under 40 and men over 60
- Onset Age: Bimodal distribution: women typically in their 20s-30s, men typically in their 60s-70s
Coding
- ICD-11: 8B44
- ICD-10-CM: G70.00
- OMIM: 254200
- UMLS: C0026896
- MeSH: D009157
- GARD: 7422
Quick Reference
| Quick Facts | Details |
|---|---|
| Incidence | 4.1-30 per million person-years (U.S.: 68.5 per million; ~17,417 new cases annually) |
| Prevalence | 150-329 per million (U.S.: 316.4 per million) |
| Gender Distribution | Bimodal: more common in women under 40 and men over 60 |
| Primary Age of Onset | Bimodal distribution: women typically 20s-30s, men typically 60s-70s |
| AT/AAC Requirements | Low to Moderate - Many benefit from AT during exacerbations or myasthenic crisis; voice amplifiers for weak speech, SGDs for severe dysarthria; fluctuating needs based on disease activity |
Medical Features and Pathophysiology
Etiology
Myasthenia Gravis (MG) is an autoimmune disorder characterized by the production of antibodies that target the acetylcholine receptors (AChR) or other components of the neuromuscular junction (NMJ). This immune response interferes with the communication between nerve cells and muscles, leading to muscle weakness and fatigue. The exact cause of this autoimmune response is unknown, but genetic and environmental factors may contribute.
Pathology
MG primarily affects the NMJ, where nerve impulses are transmitted to muscles. In most cases, antibodies attack the AChR on muscle cells, reducing the number of available receptors and impairing muscle contraction. Some patients may have antibodies against other proteins, such as muscle-specific kinase (MuSK). The reduction in effective neuromuscular transmission leads to the characteristic symptoms of muscle weakness and fatigue, which can fluctuate in intensity.
Symptoms
- Early Symptoms:
- Ptosis (drooping eyelids)
- Diplopia (double vision)
- Weakness in facial muscles
- Difficulty swallowing (dysphagia)
- Slurred speech (dysarthria)
- Progressive Symptoms:
- Generalized muscle weakness, especially after activity
- Difficulty breathing due to weakness of respiratory muscles
- Weakness in arms and legs
- Myasthenic crisis (severe muscle weakness causing respiratory failure)
Diagnosis
Diagnosis of MG involves clinical evaluation, including a detailed medical history and physical examination. Specific tests include:
- Antibody Tests: Detection of anti-AChR and anti-MuSK antibodies in the blood.
- Electrophysiological Studies: Repetitive nerve stimulation and single-fiber electromyography (SFEMG) to assess neuromuscular transmission.
- Edrophonium Test: Temporary improvement in muscle strength following administration of edrophonium, an acetylcholinesterase inhibitor.
- Imaging: CT or MRI to detect thymomas, which are associated with MG.
Assistive Suggestions and Requirements
Requirement Percentage for Assistive Technology
Many patients with MG will benefit from assistive technology, particularly during periods of exacerbation or myasthenic crisis.
Assistive Technology Suggestions
- Mobility Aids:
- Canes or Walkers: To assist with balance and mobility during periods of muscle weakness.
- Wheelchairs: For severe cases or during myasthenic crisis.
- Communication Aids:
- Voice Amplifiers: To assist with weak speech.
- Speech Generating Devices (SGDs): For severe dysarthria.
- Home Modifications:
- Grab Bars and Handrails: To enhance safety and prevent falls.
- Adjustable Beds: To improve comfort and ease of movement.
- Feeding Aids:
- Adaptive Utensils: For individuals with reduced hand strength.
Access Modalities
- Switch Access: For individuals with severe motor impairments to control communication devices and computers.
- Voice-Controlled Systems: Beneficial for those who retain good vocal strength.
- Touchscreen Devices: Useful for those with adequate hand dexterity.
Care Management and Therapeutic Techniques
Aims
- To manage symptoms and maintain the highest possible level of independence and quality of life.
- To provide supportive care and address complications through a multidisciplinary approach.
- To offer education and support to patients and caregivers.
SLP Suggestions
- Assessment and Intervention:
- Regular Speech and Swallowing Assessments: To monitor changes and adjust therapy plans accordingly.
- Swallowing Techniques: Teaching techniques to ensure safe swallowing and reduce the risk of aspiration.
- Voice Therapy: Exercises to maintain voice strength and clarity.
- Augmentative and Alternative Communication (AAC):
- Introduction of AAC devices early in the disease progression to ensure familiarity and ease of use as the disease progresses.
Special Educator Suggestions
- Cognitive Rehabilitation:
- Activities designed to enhance executive function, memory, and attention.
- Use of memory aids, such as notebooks or electronic organizers.
- Behavioral Strategies:
- Structured routines to manage apathy and maintain engagement in activities.
- Positive reinforcement to encourage participation and effort in tasks.
Occupational Therapist Suggestions
- Daily Living Skills:
- Training in the use of adaptive equipment for self-care activities (e.g., dressing, grooming).
- Techniques to conserve energy and manage fatigue.
- Home and Environmental Modifications:
- Assessing and modifying the home environment to ensure safety and accessibility.
- Recommendations for ergonomic furniture and tools to support independence.
Recommendations on AAC and Other Details
- Text-Based AAC:
- Use of text-to-speech apps and devices for individuals who retain good literacy skills.
- Predictive text features to speed up communication.
- Symbol-Based AAC:
- For individuals with cognitive impairments affecting literacy, symbol-based systems like Picture Communication Symbols (PCS) can be useful.
- Dynamic display devices that can grow with the user’s needs.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Autoimmune
- Pathology: Ideopathic
- Rarity Classification: Medium
- Typical Onset: Any Age
- Gender Impact: Either Gender
- Seizure Prevalence: No (<10%)
- Population Trend: Increasing
Pathophysiology
"Grave weakness;" error in nerve impulse transmission to muscles (PNS). Origins typically unknown, and even duration of impact can be variable.
AAC Considerations
Recommended Access Modalities: Touch,Switch
Additional Clinical Notes
Autoimmune disorder causing chronic fatigue with variable severity
Patient Advocacy & Support Organizations
References
- alchalabi. (2012).
- alasmi. (2012).
- https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Myasthenia-Gravis-Fact-Sheet
Additional Information
MG requires ongoing neurological management with regular assessment of treatment efficacy and disease activity. Monitoring for myasthenic crisis and respiratory complications is essential, particularly during infections or medication changes. Coordination between neurology, pulmonology, and rehabilitation services optimizes functional outcomes and quality of life.
Extended Information
Pathological Insights and Disease Mechanism
Myasthenia Gravis (MG) is characterized by the production of autoantibodies that disrupt neuromuscular transmission, leading to muscle weakness and fatigue. In most patients, these antibodies target the acetylcholine receptor (AChR) at the neuromuscular junction. A smaller subset of patients has antibodies against muscle-specific kinase (MuSK) or other components of the neuromuscular junction. The presence of these autoantibodies reduces the number of functional AChRs, impairs synaptic transmission, and ultimately weakens muscle contractions. The exact triggers for the autoimmune response are not fully understood, but genetic predisposition, infections, and other environmental factors are thought to play a role.
Genetic and Environmental Factors
While MG is primarily an autoimmune disorder, genetic factors may contribute to the susceptibility to developing the disease. Certain genetic polymorphisms in immune-related genes have been associated with an increased risk of MG. Environmental factors, such as viral infections, have also been implicated in triggering the autoimmune response. The interplay between genetic predisposition and environmental triggers is an area of active research, aiming to uncover the underlying mechanisms that lead to the development of MG.
Clinical Presentation and Disease Progression
The clinical presentation of MG varies widely among individuals. The hallmark symptoms include fluctuating muscle weakness and fatigue, which worsen with activity and improve with rest. The disease often begins with ocular symptoms, such as ptosis (drooping eyelids) and diplopia (double vision), which may progress to generalized muscle weakness. In some cases, MG can lead to severe respiratory muscle weakness, resulting in myasthenic crisis—a life-threatening condition that requires immediate medical attention. The course of the disease can be variable, with some patients experiencing periods of remission and others having a progressive decline in muscle strength.
Differential Diagnosis and Overlapping Syndromes
Diagnosing MG involves differentiating it from other neuromuscular disorders that can present with similar symptoms. Conditions such as Lambert-Eaton Myasthenic Syndrome (LEMS), botulism, and primary muscle diseases need to be considered. A thorough clinical evaluation, including a detailed history, physical examination, and appropriate diagnostic tests, is essential for an accurate diagnosis. Electrophysiological studies and antibody testing are particularly important for confirming MG and ruling out other conditions.
Therapeutic Interventions and Symptom Management
The management of MG involves a combination of pharmacological and non-pharmacological treatments aimed at improving neuromuscular transmission, suppressing the autoimmune response, and managing symptoms. Acetylcholinesterase inhibitors, such as pyridostigmine, are commonly used to enhance neuromuscular transmission and alleviate muscle weakness. Immunosuppressive therapies, including corticosteroids, azathioprine, and mycophenolate mofetil, are used to reduce
the production of autoantibodies and modulate the immune response. Intravenous immunoglobulin (IVIG) and plasmapheresis rapidly reduce antibody levels during myasthenic crises or before surgery. Thymectomy is recommended for individuals with thymoma or generalized MG, potentially leading to long-term remission.
Research and Future Directions
Research focuses on understanding autoimmune mechanisms and developing targeted immunotherapies. Monoclonal antibodies targeting specific immune system components show promise in clinical trials. Biomarker development aims to improve early diagnosis and disease monitoring.
Support and Resources
Organizations such as the Myasthenia Gravis Foundation of America (MGFA) and Myaware provide educational materials, support networks, and research advocacy. Support groups offer platforms for sharing experiences and accessing practical guidance.
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Myasthenia Gravis?
What does Myasthenia Gravis do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.