Introduction
Pompe disease results from mutations in the GAA gene, causing deficiency of acid alpha-glucosidase enzyme. This leads to pathological glycogen accumulation in lysosomes, particularly affecting cardiac and skeletal muscle. Clinical presentation varies by residual enzyme activity. Infantile-onset disease presents with severe hypotonia, cardiomegaly, and respiratory failure, typically within the first months of life. Late-onset forms manifest with progressive proximal muscle weakness and respiratory insufficiency, beginning anywhere from early childhood to adulthood. Enzyme replacement therapy with alglucosidase alfa has transformed outcomes, particularly for infantile-onset cases.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Genetic
- Pathology: Heritable
- Rarity Classification: Rare
- Typical Onset: Birth to Adolescence
- Gender Impact: Either Gender
- Progressive/Degenerative: Yes
- Seizure Prevalence: Variable (Subtype)
- Population Trend: Unknown
Pathophysiology
Autosomal recessive mutation of the GAA gene, causing impaired ability to break down sugars leading to accumulations of glucose in bodily tissues.
AAC Considerations
Recommended Access Modalities: All - Progression
Patient Advocacy & Support Organizations
- http://www.amda-pompe.org/
- https://www.agsdus.org/type-ii.php
- https://agsd.org.uk/
- https://worldpompe.org/
Pompe Disease Handout
Formal Name: Glycogen Storage Disease Type II
Demographic Information
- Incidence: Approximately 1 in 40,000 births worldwide
- Prevalence: Varies by population, with some higher incidences in certain ethnic groups
- Gender: Affects both males and females equally
- Onset Age: Can manifest at any age, with three main forms: infantile-onset, late-onset juvenile, and late-onset adult
Coding
- ICD-11: 5C50.0
- ICD-10-CM: E74.02
- OMIM: 232300
- UMLS: C0032580
- MeSH: D006009
- GARD: 7197
Medical Features and Pathophysiology
Etiology
Pompe disease is a genetic disorder caused by mutations in the GAA gene, which encodes the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen into glucose within lysosomes. Mutations in the GAA gene lead to deficient or absent GAA enzyme activity, resulting in the accumulation of glycogen in various tissues, particularly muscles.
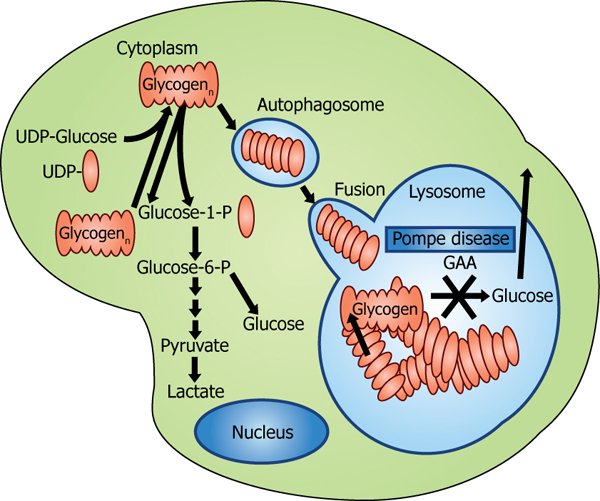
Pathology
The accumulation of glycogen in lysosomes causes damage to muscle cells, leading to progressive muscle weakness and loss of function. The severity and progression of the disease depend on the level of residual GAA enzyme activity. Infantile-onset Pompe disease is the most severe form, presenting within the first few months of life with rapid progression, while late-onset forms have a more variable and slower progression.
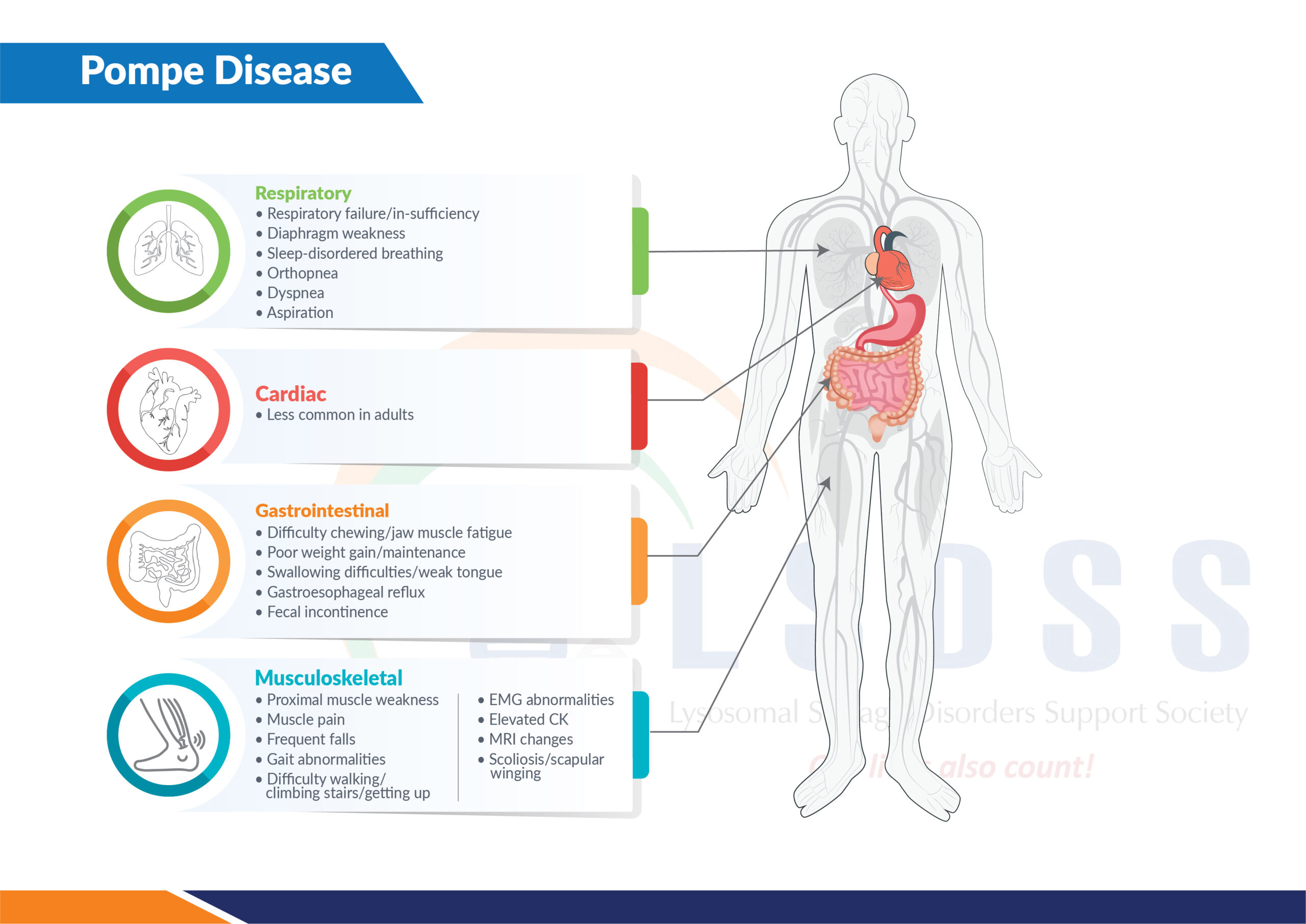
Symptoms
- Infantile-Onset:
- Hypotonia (poor muscle tone)
- Cardiomegaly (enlarged heart)
- Hepatomegaly (enlarged liver)
- Respiratory difficulties
- Feeding problems and failure to thrive
- Late-Onset:
- Progressive muscle weakness, especially in the legs and trunk
- Respiratory muscle weakness leading to breathing difficulties
- Fatigue and exercise intolerance
- Muscle cramps and pain
Diagnosis
Diagnosis of Pompe disease involves several steps:
- Enzyme Assay: Measurement of GAA enzyme activity in blood, skin fibroblasts, or muscle tissue.
- Genetic Testing: Identification of mutations in the GAA gene.
- Electromyography (EMG): To assess muscle function.
- Muscle Biopsy: To observe glycogen accumulation and muscle pathology.
- Cardiac Evaluation: Particularly important for infantile-onset cases to assess heart function.
Assistive Suggestions and Requirements
Requirement Percentage for Assistive Technology
Most patients with Pompe disease will benefit from assistive technology, especially as the disease progresses and muscle weakness increases.
Assistive Technology Suggestions
Mobility Aids:
- Wheelchairs and Scooters: For individuals with significant mobility impairment.
- Canes and Walkers: To support those with less severe weakness.
Respiratory Aids:
- Non-invasive Ventilation (NIV): To assist with breathing, especially during sleep.
- Cough Assist Devices: To help clear airway secretions.
Home Modifications:
- Stair Lifts and Ramps: To improve accessibility at home.
- Grab Bars and Handrails: For safety and support.
Communication Aids:
- Speech Generating Devices (SGDs): For individuals with severe respiratory muscle weakness affecting speech.
Access Modalities
- Switch Access: For those with severe motor impairments.
- Voice-Controlled Systems: Beneficial for those with adequate vocal strength.
- Touchscreen Devices: For those with sufficient hand function.
Care Management and Therapeutic Techniques
Aims
- To manage symptoms and enhance quality of life.
- To provide multidisciplinary care involving neurologists, cardiologists, pulmonologists, physical therapists, and genetic counselors.
- To offer ongoing support and education to patients and caregivers.
SLP Suggestions
Assessment and Intervention:
- Regular Speech and Swallowing Assessments: To monitor and address dysphagia and dysarthria.
- Swallowing Techniques: Training to ensure safe swallowing and reduce aspiration risk.
- Voice Therapy: Exercises to maintain voice strength.
Augmentative and Alternative Communication (AAC):
- Early introduction of AAC devices to facilitate communication.
Special Educator Suggestions
Cognitive and Behavioral Strategies:
- Structured routines to support engagement in educational activities.
- Use of memory aids and electronic organizers to enhance cognitive function.
Occupational Therapist Suggestions
Daily Living Skills:
- Training in the use of adaptive equipment for activities of daily living.
- Techniques for energy conservation and fatigue management.
Home and Environmental Modifications:
- Recommendations for ergonomic furniture and tools to support independence.
- Safety assessments and modifications to the home environment.
Recommendations on AAC and Other Details
Text-Based AAC:
- Utilization of text-to-speech applications for those with preserved literacy skills.
- Predictive text features to facilitate communication.
Symbol-Based AAC:
- Use of symbols for individuals with cognitive impairments affecting literacy.
Additional Information
Pompe disease requires lifelong multidisciplinary management. Regular monitoring includes pulmonary function testing, cardiac evaluation, and assessment of motor function. Physical therapy maintains strength and prevents contractures. Respiratory support, initially with non-invasive ventilation during sleep, often progresses to full-time ventilatory support in advanced disease. Nutritional management addresses feeding difficulties and maintains adequate caloric intake.
Genetic counseling helps families understand autosomal recessive inheritance patterns and recurrence risks. Newborn screening programs in many regions enable early diagnosis and initiation of enzyme replacement therapy, significantly improving outcomes for infantile-onset cases.
Extended Information
Pathological Insights and Disease Mechanism
Pompe disease is characterized by the accumulation of glycogen in lysosomes due to a deficiency of the enzyme acid alpha-glucosidase (GAA). This accumulation disrupts normal cellular function, particularly in muscle cells, leading to progressive muscle weakness and other systemic complications. Infantile-onset Pompe disease presents with severe symptoms shortly after birth, including profound muscle weakness, cardiomegaly, and respiratory distress. Late-onset forms present with a more gradual progression of muscle weakness and respiratory involvement.
Genetic and Environmental Factors
Pompe disease is inherited in an autosomal recessive pattern, meaning that an individual must inherit two mutated copies of the GAA gene to develop the disease. Carriers, who have one normal and one mutated gene, typically do not show symptoms but can pass the mutation to their offspring. Genetic counseling is recommended for families affected by Pompe disease to understand the inheritance pattern and assess the risk of recurrence in future pregnancies.
Clinical Presentation and Disease Progression
The clinical presentation of Pompe disease varies based on the age of onset and severity of enzyme deficiency. Infantile-onset Pompe disease is the most severe form, with rapid progression and life-threatening complications if untreated. Late-onset Pompe disease progresses more slowly, with symptoms such as muscle weakness, respiratory difficulties, and fatigue becoming more pronounced over time. Early diagnosis and intervention are crucial to managing the disease and improving outcomes.
Differential Diagnosis and Overlapping Syndromes
Differentiating Pompe disease from other neuromuscular disorders is essential for accurate diagnosis and appropriate treatment. Conditions such as muscular dystrophies, spinal muscular atrophy, and other glycogen storage diseases can present with similar symptoms. Comprehensive diagnostic evaluations, including enzyme assays, genetic testing, and muscle biopsies, are necessary to confirm Pompe disease and exclude other conditions.
Therapeutic Interventions and Symptom Management
The primary treatment for Pompe disease is enzyme replacement therapy (ERT) with alglucosidase alfa, which provides the deficient GAA enzyme and helps reduce glycogen accumulation. ERT has been shown to improve muscle strength, respiratory function, and overall survival, particularly in infantile-onset cases. Supportive treatments, such as physical therapy, respiratory therapy, and dietary modifications, are also important for managing symptoms and improving quality of life. Regular monitoring and adjustments to the treatment plan are necessary to address the evolving needs of individuals with Pompe disease.
Research and Future Directions
Research into Pompe disease continues to explore new therapeutic approaches, including gene therapy, which aims to provide a long-term cure by delivering a functional copy of the GAA gene to patients' cells. Other areas of research focus on improving the efficacy of ERT, developing small-molecule therapies to enhance enzyme activity, and identifying biomarkers for early diagnosis and disease monitoring. Collaborative efforts among researchers, clinicians, and patient advocacy groups are essential to advancing the understanding and treatment of Pompe disease.
Support and Resources
Organizations such as the Acid Maltase Deficiency Association (AMDA) and the International Pompe Association (IPA) provide valuable resources, support networks, and advocacy for individuals affected by Pompe disease. These organizations offer educational materials, connect patients and families with healthcare professionals, and fund research initiatives aimed at finding a cure and improving the quality of life for those living with Pompe disease. Support groups, both in-person and online, provide a platform for patients and caregivers to share experiences, receive emotional support, and access practical advice.
Gene therapy approaches aim to provide sustained GAA expression, potentially offering long-term treatment alternatives to biweekly enzyme infusions. Research also explores chaperone therapies to enhance residual enzyme activity and novel enzyme formulations with improved tissue penetration.
References
- Kishnani, P. S., & Howell, R. R. (2004). Pompe disease in infants and children. Journal of Pediatrics, 144(5 Suppl), S35-S43.
- van der Ploeg, A. T., & Reuser, A. J. J. (2008). Pompe's disease. The Lancet, 372(9646), 1342-1353.
- van Capelle, C. I., et al. (2016). Efficacy of enzyme replacement therapy in juvenile patients with Pompe disease: a systematic review. Journal of Neurology, 263(5), 855-870.
- https://ghr.nlm.nih.gov/condition/pompe-disease
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Pompe Disease?
What does Pompe Disease do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.