Introduction
Prader-Willi syndrome is a common rare genetic disorder affecting both genders equally, with symptoms present from birth. It is caused by the absence of expression of paternally inherited genes on chromosome 15q11-q13, impacting the hypothalamus and resulting in symptoms like poor muscle tone, excessive hunger, developmental delays, and endocrine abnormalities. Diagnosis is based on clinical features and genetic testing. Management requires assistive technology, visual schedules, sensory integration tools, and a multidisciplinary approach to manage hyperphagia and obesity, support cognitive and social development, and improve the overall quality of life.
Clinical Information
The following data is from validated clinical sources and patient registries.
Core Characteristics
- Etiology: Genetic
- Pathology: Mutation - Somatic
- Rarity Classification: Rare
- Typical Onset: Birth
- Gender Impact: Either Gender
- Seizure Prevalence: Yes (>90%)
- Population Trend: Stable
Pathophysiology
Deletion of genes on Chromosome 15 in either paternal or maternal sex cells during their development - or very rarely, translocation of those genes. About 70% of cases are associated with a mutation in a single chromosome on the paternal side, with the other 30% occuring via "uniparental disomy" where both chromosomes come from either the mother or father. Genetically very interesting, RNA clusters and the OCA2 gene are also sometimes involved; further investigation recommended to anyone interested but will leave this summary there.
AAC Considerations
Recommended Access Modalities: Touch
Additional Clinical Notes
Gene deletion due to chromosomal anomaly in the father; constant hunger and weak musculature are hallmarks but intellectual disability is frequent. This may actually be a bigger addressable market than the numbers indicate here, I'm being very conservative, but it appears to be wildly underdiagnosed
Patient Advocacy & Support Organizations
Prader-Willi Syndrome
Demographic Information
- Incidence: Prader-Willi syndrome (PWS) occurs in approximately 1 in 10,000 to 30,000 live births, making it one of the more common rare genetic disorders.
- Prevalence: The global prevalence of Prader-Willi syndrome is estimated to be around 1 in 20,000 to 1 in 25,000 people. This prevalence can vary slightly due to diagnostic criteria and awareness levels in different regions.
- Gender: Prader-Willi syndrome affects both males and females equally. There is no gender predilection for this disorder.
- Onset Age: Symptoms of Prader-Willi syndrome can be present from birth, with hypotonia (poor muscle tone) being a prominent early sign. However, the hallmark features such as hyperphagia (excessive hunger) and behavioral problems typically become more apparent in early childhood.
Coding
- ICD-11: LD90.3
- ICD-10-CM: Q87.19
- OMIM: 176270 (Prader-Willi Syndrome)
- UMLS: C0032897 (Prader-Willi Syndrome)
- MeSH: D011339 (Prader-Willi Syndrome)
- GARD: 7778 (Prader-Willi Syndrome)
Medical Features and Pathophysiology
Etiology
Prader-Willi syndrome is a complex genetic disorder caused by the absence of expression of paternally inherited genes on chromosome 15q11-q13. The specific mechanisms leading to this lack of gene expression include:
- Paternal Deletion: The most common cause, occurring in about 70% of cases, where a segment of the paternal chromosome 15 is deleted.
- Maternal Uniparental Disomy (UPD): In about 25% of cases, both copies of chromosome 15 are inherited from the mother, leading to a lack of paternal gene expression.
- Imprinting Defects: These account for the remaining cases and involve errors in the genomic imprinting process, which affects the regulation of genes in the Prader-Willi critical region.
Pathology
The absence of expression of these crucial paternal genes affects the hypothalamus, a region of the brain that plays a key role in regulating hunger, growth, metabolism, and other endocrine functions. This disruption results in the characteristic features and symptoms of Prader-Willi syndrome, which include hypotonia, hyperphagia, developmental delays, and endocrine abnormalities.
Symptoms
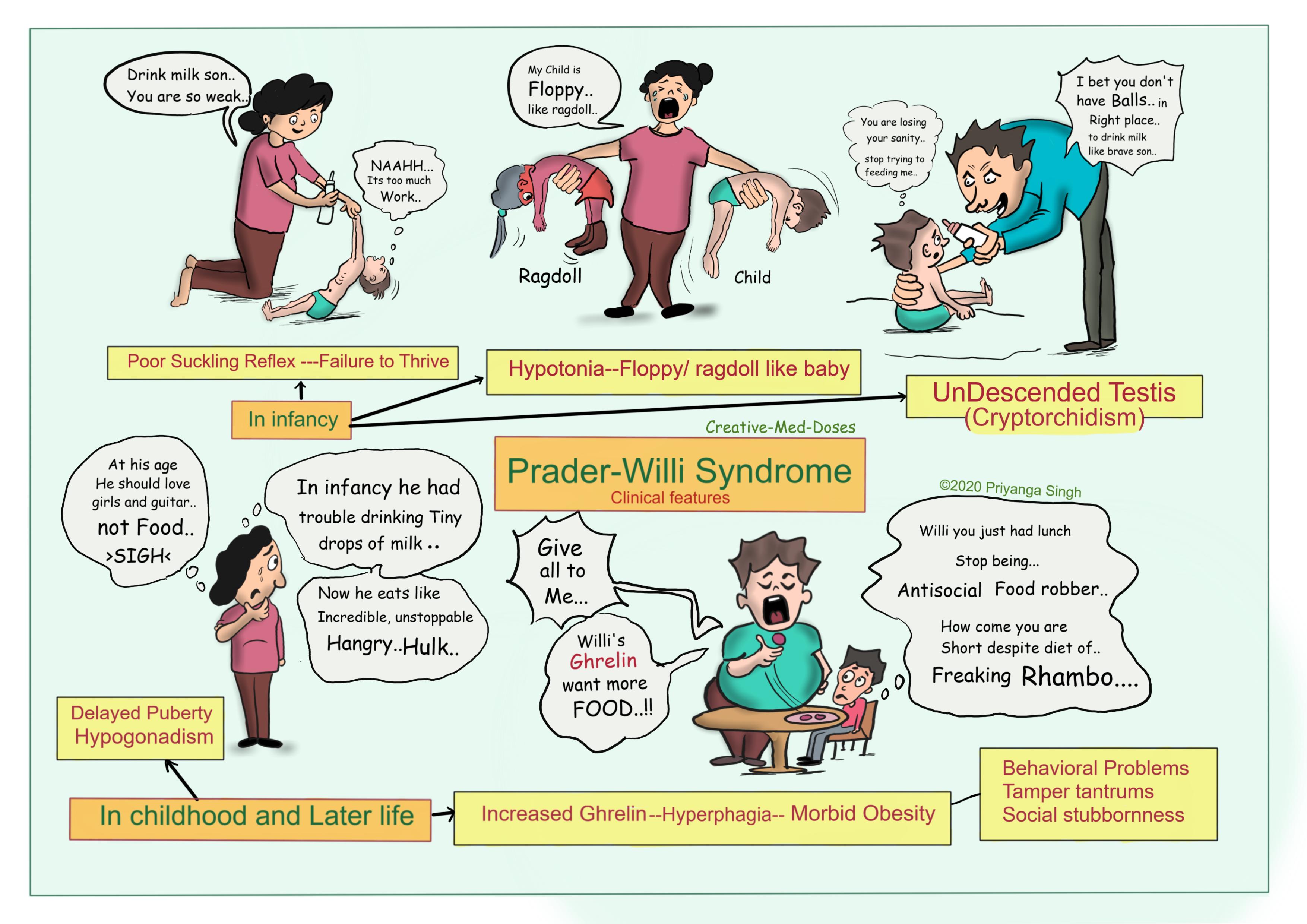
- Neonatal Period: Common signs in infancy include severe hypotonia, which leads to poor sucking and feeding difficulties, often requiring special feeding techniques or gastrostomy.
- Childhood: As children with Prader-Willi syndrome grow, they develop an insatiable appetite, leading to hyperphagia and rapid weight gain if food intake is not strictly controlled. Developmental delays and intellectual disabilities are also prominent during this period.
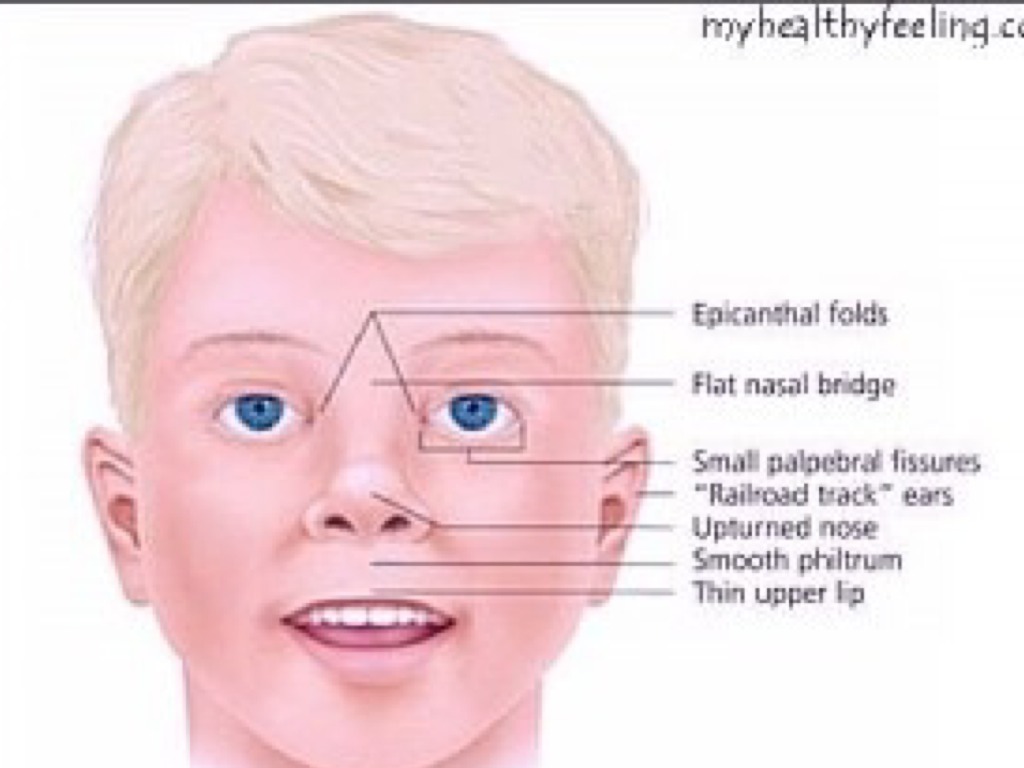
- Physical Features: Characteristic facial features include a narrow forehead, almond-shaped eyes, and a triangular mouth. Other physical features may include small hands and feet, short stature, and scoliosis.
- Behavioral Issues: Behavioral problems such as temper tantrums, stubbornness, and compulsive behaviors like skin picking are common. These behaviors can be challenging to manage and often require behavioral interventions.
- Endocrine Issues: Hypogonadism, resulting in incomplete sexual development and infertility, is common. Growth hormone deficiency, which can contribute to short stature and reduced muscle mass, is also frequently observed.
- Sleep Disorders: Sleep apnea and other sleep-related issues, such as excessive daytime sleepiness, are prevalent in individuals with Prader-Willi syndrome.
Diagnosis
The diagnosis of Prader-Willi syndrome is based on a combination of clinical features and genetic testing. Genetic tests such as methylation analysis, fluorescence in situ hybridization (FISH), and chromosomal microarray can identify the specific genetic abnormalities causing the disorder.
Assistive Suggestions and Requirements
Requirement Percentage for Assistive Technology
Most individuals with Prader-Willi syndrome require assistive technology to support dietary management, communication, and mobility.
Assistive Technology Suggestions
- Feeding and Nutritional Management: Tools such as food diaries, calorie counters, and specialized kitchen appliances can help manage hyperphagia. Environmental controls like locked cabinets and refrigerators are essential to prevent overeating.
- Communication Devices: Augmentative and alternative communication (AAC) devices can support those with speech delays or impairments. These devices range from simple picture boards to advanced speech-generating devices, helping individuals express their needs and interact with others.
- Mobility Aids: Depending on the individual's motor skills and physical condition, mobility aids such as orthotic devices, wheelchairs, or walkers may be necessary to support movement and prevent obesity-related complications.
Access Modalities
- Visual Schedules: Visual schedules and organizational tools help individuals with Prader-Willi syndrome understand daily routines and reduce anxiety. These can include pictorial calendars, apps, or physical boards.
- Sensory Integration Tools: Tools such as weighted blankets, sensory rooms, and fidget devices can help manage sensory processing issues and behavioral problems, providing a calming effect and improving focus.
Care Management and Therapeutic Techniques
Aims
The primary aims of care management for individuals with Prader-Willi syndrome are to manage hyperphagia and obesity, support cognitive and social development, address behavioral issues, and improve overall quality of life through a multidisciplinary approach.
SLP (Speech-Language Pathologist) Suggestions
- Language and Communication Therapy: Focus on developing speech and language skills through structured activities, social interactions, and the use of AAC devices. Consistent practice and reinforcement are key to improving communication abilities.
- Feeding Therapy: Address early feeding difficulties with techniques to improve muscle tone and coordination for effective swallowing. For older children, provide guidance on healthy eating habits and portion control to manage hyperphagia.
- Social Skills Training: Encourage social interactions through group activities, role-playing, and other methods to develop pragmatic language skills and reduce social isolation.
Special Educator Suggestions
- Individualized Education Plan (IEP): Develop an IEP tailored to the student's cognitive and behavioral needs. Goals should focus on academic achievement, social skills, and independence.
- Behavioral Interventions: Implement behavior management strategies to address temper tantrums, stubbornness, and compulsive behaviors. Positive reinforcement, clear expectations, and structured environments are effective in managing these behaviors.
- Functional Skills Training: Emphasize the development of life skills, such as self-care, vocational training, and adaptive skills, to promote independence and self-sufficiency.
Occupational Therapist Suggestions
- Fine and Gross Motor Skills: Use exercises and activities to improve muscle tone, coordination, and strength. Tailored interventions can help with fine motor skills needed for daily tasks and improve overall physical abilities.
- Sensory Integration Therapy: Provide sensory integration therapy to help manage sensory processing issues. This can include activities that stimulate the senses in a controlled manner, promoting regulation and reducing behavioral problems.
- Daily Living Skills: Train in activities of daily living (ADLs), including personal hygiene, dressing, and cooking, using adaptive tools as needed. Encourage participation in community and recreational activities to build social skills and independence.
Recommendations on AAC and Other Details
AAC systems for individuals with Prader-Willi syndrome benefit from careful customization matched to cognitive and motor abilities. Symbol-based tools often form the foundation, given the visual strengths many individuals demonstrate.
- Customized AAC Systems: Personalize AAC systems and update regularly. Training for caregivers, educators, and therapists ensures consistent communication support.
- Symbol-Based Communication: Picture exchange systems, visual schedules, and symbol-supported literacy activities should use clear, consistent symbols tailored to comprehension level.
- Collaborative Approach: Maintain consistent communication strategies across families, educators, and therapists. Regular team meetings align interventions. Active family involvement in decision-making strengthens implementation.
References
- Cassidy, S. B., Schwartz, S., Miller, J. L., & Driscoll, D. J. (2012). Prader-Willi syndrome. Genetics in Medicine, 14(1), 10-26.
- Butler, M. G., Hartin, S. N., Hossain, W. A., Manzardo, A. M., Kimonis, V., Dykens, E., Gold, J. A., Kim, S. J., & Weisensel, N. (2019). Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with Prader-Willi syndrome. American Journal of Medical Genetics Part A, 179(8), 1407-1414.
- Goldstone, A. P., Holland, A. J., Hauffa, B. P., Hokken-Koelega, A. C., & Tauber, M. (2008). Recommendations for the diagnosis and management of Prader-Willi syndrome. Journal of Clinical Endocrinology & Metabolism, 93(11), 4183-4197.
- Miller, J. L., Lynn, C. H., Driscoll, D. C., Goldstone, A. P., Gold, J. A., Kimonis, V., Dykens, E., Butler, M. G., Shuster, J. J., & Driscoll, D. J. (2011). Nutritional phases in Prader-Willi syndrome. American Journal of Medical Genetics Part A, 155A(5), 1040-1049.
- Tauber, M.,
& Hoybye, C. (2020). Endocrine disorders in Prader-Willi syndrome: A model to understand and treat hypothalamic dysfunction. Lancet Diabetes & Endocrinology, 8(7), 552-561.
In conclusion, Prader-Willi syndrome is a complex genetic disorder that requires a comprehensive and multidisciplinary approach to management. Early diagnosis, targeted interventions, and ongoing support are essential for improving the quality of life for individuals with Prader-Willi syndrome. Advances in research and therapeutic strategies continue to offer hope for better management and understanding of this challenging condition.
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Prader-Willi?
What does Prader-Willi do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.