Introduction
Table of Contents
Coding
- ICD-11: LD90.4
- ICD-10-CM: F84.2
Quick Reference
| Quick Facts | Details |
|---|---|
| Incidence | 1 in 10,000 to 15,000 live female births |
| Prevalence | 1 in 10,000 to 23,000 females globally; predominantly affects females (X-linked) |
| Gender Distribution | Almost exclusively females; extremely rare in males (often fatal early) |
| Primary Age of Onset | Symptoms appear 6-18 months; regression stage 1-4 years; diagnosis average 2.7 years |
| AT/AAC Requirements | Critical - Profound loss of purposeful hand movements and speech requires AAC for virtually all |
Introduction
Rett syndrome is a rare neurodevelopmental disorder that primarily affects females, with an incidence of approximately 1 in 10,000 to 15,000 live female births. This disorder is characterized by apparently normal early development followed by a profound regression of acquired skills, particularly in the domains of purposeful hand movements and spoken language. Rett syndrome is caused by mutations in the MECP2 gene, which is located on the X chromosome and encodes the methyl-CpG-binding protein 2 (MeCP2), a protein essential for normal brain development and function.
This comprehensive guide aims to provide healthcare professionals with an in-depth understanding of Rett syndrome, encompassing its epidemiology, etiology, pathophysiology, clinical features, diagnostic criteria, and management strategies. Special emphasis is placed on communication challenges, assistive technology interventions, and augmentative and alternative communication (AAC) approaches, as these are crucial for enhancing the quality of life for individuals with Rett syndrome.
Epidemiology and Demographics
The global prevalence of Rett syndrome is estimated to be between 1 in 10,000 to 23,000 females, with slight variations across different populations due to differences in diagnostic criteria and potential underdiagnosis in certain regions. The disorder predominantly affects females due to its genetic link to the X chromosome. Males can also be affected, but it is extremely rare, and they often experience more severe outcomes and early mortality. In males, Rett syndrome-like features may be part of a broader spectrum of neurodevelopmental disorders caused by MECP2 mutations, such as MECP2 duplication syndrome.
The onset of symptoms typically occurs between 6 to 18 months of age, following a period of seemingly normal early development. During this early period, infants with Rett syndrome may achieve typical developmental milestones such as sitting, crawling, and even walking and babbling. However, parents often notice a slowing of development or a loss of previously acquired skills between 1 to 4 years of age, marking the onset of the regression stage.
The average age of diagnosis is around 2.7 years, although earlier diagnosis is becoming more common with increased awareness and improved genetic testing. Early diagnosis is crucial for initiating appropriate interventions and support services, which can significantly improve outcomes and quality of life for individuals with Rett syndrome and their families.
Causes and Impact
What causes Rett Syndrome?
Etiology and Pathophysiology
Rett syndrome is primarily caused by mutations in the MECP2 gene, which is located on the X chromosome at Xq28. This gene encodes the methyl-CpG-binding protein 2 (MeCP2), a multifunctional protein that plays a crucial role in regulating the expression of other genes essential for normal brain development and function. MeCP2 acts as a transcriptional repressor, binding to methylated DNA and recruiting other proteins to modify chromatin structure and control gene expression.
Over 200 different mutations in the MECP2 gene have been identified in individuals with Rett syndrome, with some mutations being more common and associated with specific clinical features or severity of the disorder. The most common mutations are missense mutations, nonsense mutations, and small insertions or deletions, which can lead to the production of a non-functional or truncated MeCP2 protein. Large deletions and complex rearrangements of the MECP2 gene have also been reported, although less frequently.
In the majority of cases, these mutations occur de novo, meaning they are not inherited from parents but instead arise spontaneously in the individual. However, in rare instances, Rett syndrome can be inherited due to germline mosaicism, where a parent carries the mutation in a subset of their germ cells (egg or sperm).
The X-linked inheritance pattern of Rett syndrome explains its predominance in females. Females have two X chromosomes, and in each cell, one of the X chromosomes is randomly inactivated (a process known as X-chromosome inactivation). This means that females with a MECP2 mutation on one X chromosome will have a mixture of cells expressing either the normal or mutant MeCP2 protein, resulting in a mosaic pattern of gene expression. The severity of Rett syndrome in females can be influenced by the specific mutation and the X-inactivation pattern, with skewed X-inactivation favoring the expression of the normal MECP2 allele associated with milder phenotypes.
In males, who have only one X chromosome, a MECP2 mutation usually results in severe neonatal encephalopathy and early lethality, as they lack a second normal copy of the gene. However, in rare cases, males with MECP2 mutations can survive and present with a range of neurodevelopmental phenotypes, including Rett syndrome-like features, depending on the specific mutation and the presence of somatic mosaicism.
What does Rett Syndrome do to the body?
It all comes down to proteins.
The MeCP2 protein is widely expressed in the brain and is essential for normal brain development, synaptic plasticity, and the regulation of various genes involved in neuronal function. In Rett syndrome, mutations in the MECP2 gene lead to a deficiency or dysfunction of the MeCP2 protein, disrupting these critical processes and leading to the neurological and developmental abnormalities characteristic of the disorder.
The absence or dysfunction of MeCP2 results in widespread disruption of gene expression in the brain. MeCP2 normally acts as a transcriptional repressor, binding to methylated DNA and recruiting co-repressor complexes to silence target genes. Loss of MeCP2 function leads to the inappropriate expression of these target genes, which can include genes involved in neuronal development, synaptic function, and neurotransmitter signaling. Additionally, MeCP2 has been shown to interact with other transcriptional regulators and chromatin-modifying enzymes, suggesting that its loss may have far-reaching effects on gene expression and chromatin structure.
The disruption of MeCP2 function also leads to altered synaptic plasticity, which is crucial for learning, memory, and the ability of the brain to adapt to new experiences. Studies in animal models of Rett syndrome have demonstrated abnormalities in synaptic structure and function, including reduced dendritic branching, altered spine density, and impaired long-term potentiation (LTP) and long-term depression (LTD). These synaptic deficits are thought to contribute to the cognitive and motor impairments observed in Rett syndrome.
Another important aspect of Rett syndrome pathophysiology is the imbalance in excitatory and inhibitory neurotransmission. Studies have shown alterations in the levels and signaling of key neurotransmitters such as glutamate (excitatory) and GABA (inhibitory) in Rett syndrome models. This imbalance can lead to neuronal hyperexcitability and may contribute to the seizures and other neurological symptoms observed in individuals with Rett syndrome.
MeCP2 also plays a role in regulating the maturation and maintenance of neurons throughout life. In Rett syndrome, the disruption of MeCP2 function can lead to impaired neuronal differentiation, reduced neuronal size, and altered neuronal connectivity. These abnormalities in neuronal development and maintenance likely contribute to the progressive nature of the disorder and the characteristic developmental regression observed in affected individuals.
Furthermore, metabolic abnormalities have been reported in individuals with Rett syndrome and in animal models of the disorder. These abnormalities include mitochondrial dysfunction, oxidative stress, and altered cholesterol metabolism. While the precise role of these metabolic changes in the pathogenesis of Rett syndrome remains to be fully elucidated, they may contribute to the energy deficits and cellular dysfunction observed in affected neurons and other tissues.
The complex interplay of these pathophysiological mechanisms - disrupted gene expression, altered synaptic plasticity, imbalanced neurotransmission, impaired neuronal development and maintenance, and metabolic abnormalities - ultimately leads to the neurodevelopmental regression and multisystemic symptoms characteristic of Rett syndrome. Understanding these underlying mechanisms is crucial for developing targeted therapeutic strategies to ameliorate the symptoms and improve the quality of life for individuals with this disorder.
Differential Diagnosis
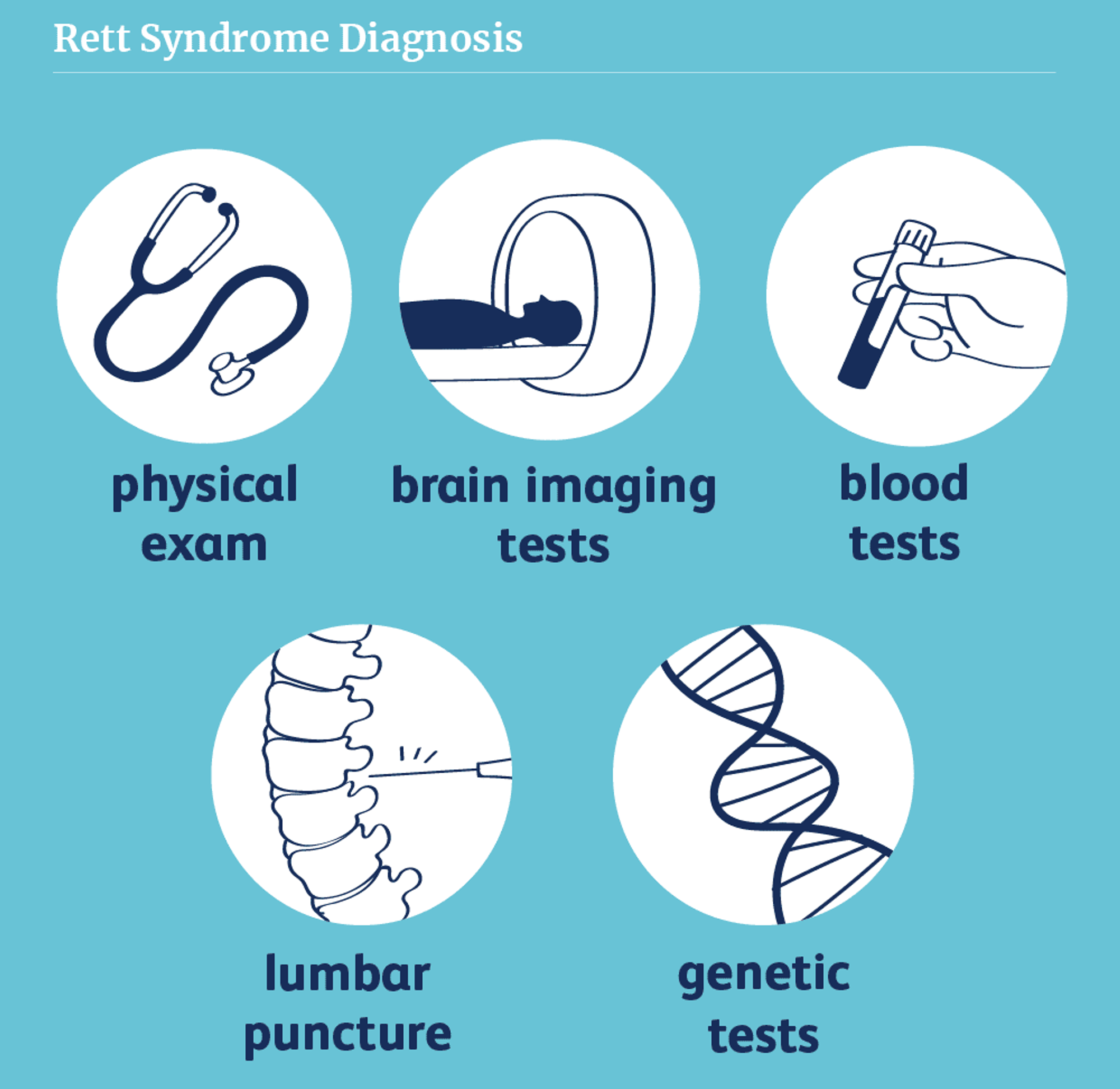
The diagnosis of Rett syndrome involves a combination of clinical assessment and genetic testing. The latest diagnostic criteria for Rett syndrome, published in 2010, emphasize the core features of the disorder and provide guidance for differentiating Rett syndrome from other neurodevelopmental conditions.
Primary Diagnostic Criteria
- A period of regression followed by recovery or stabilization: This is the hallmark of Rett syndrome and is essential for the diagnosis. The regression typically occurs between 1 to 4 years of age and involves the loss of previously acquired hand skills and spoken language.
- All main criteria:
- Partial or complete loss of acquired purposeful hand skills
- Partial or complete loss of acquired spoken language
- Gait abnormalities: Impaired (dyspraxic) or absent ability to walk
- Stereotypic hand movements such as hand wringing/squeezing, clapping/tapping, mouthing, and washing/rubbing automatisms.
- Supportive criteria are not required for diagnosis but may be present:
- Breathing disturbances when awake
- Bruxism (teeth grinding) when awake
- Impaired sleep pattern
- Abnormal muscle tone
- Peripheral vasomotor disturbances
- Scoliosis/kyphosis
- Growth retardation
- Small cold hands and feet
- Inappropriate laughing/screaming spells
- Diminished response to pain
- Intense eye communication ("eye pointing")
Exclusion Criteria
- Brain injury secondary to trauma (peri- or postnatally), neurometabolic disease, or severe infection that causes neurological problems
- Grossly abnormal psychomotor development in the first 6 months of life
The presence of any of these exclusion criteria should prompt consideration of alternative diagnoses or further investigation to determine if the child has a Rett-like disorder with a different genetic basis.
Genetic Testing
Useful, but not always reliable!
Genetic testing is a crucial component of the diagnostic process for Rett syndrome. Approximately 95-97% of individuals who meet the clinical diagnostic criteria for typical Rett syndrome have a detectable mutation in the MECP2 gene. Genetic testing involves sequencing the MECP2 gene to identify pathogenic variants, which can include missense mutations, nonsense mutations, insertions, deletions, and large rearrangements.
However, it is important to note that a small percentage of individuals with clinically diagnosed Rett syndrome (3-5%) do not have detectable MECP2 mutations. In these cases, further genetic testing may be warranted to investigate other genes associated with Rett-like phenotypes, such as CDKL5 and FOXG1. Mutations in these genes can cause distinct but overlapping neurodevelopmental disorders with some features reminiscent of Rett syndrome.
Genetic testing is not only valuable for confirming the diagnosis of Rett syndrome but also for providing information about the specific mutation, which can help guide prognosis, genetic counseling, and potential eligibility for future mutation-specific therapies. Additionally, identifying the genetic basis of the disorder can provide closure and support for families, who often experience a prolonged diagnostic odyssey before receiving a definitive diagnosis.
In summary, the diagnosis of Rett syndrome involves a comprehensive clinical evaluation to identify the characteristic features and developmental trajectory of the disorder, combined with genetic testing to detect pathogenic variants in the MECP2 gene or other associated genes. A timely and accurate diagnosis is essential for accessing appropriate medical care, support services, and educational interventions that can significantly improve the quality of life for individuals with Rett syndrome and their families.
Clinical Features and Stages
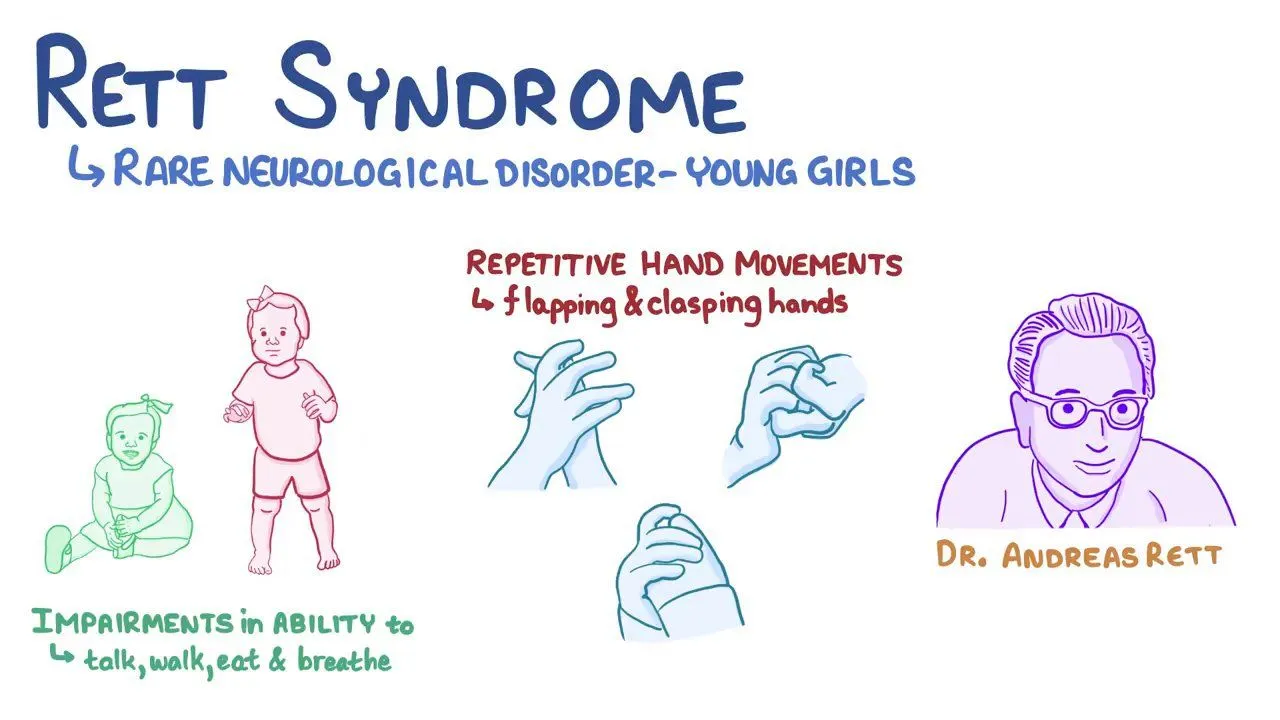
Rett syndrome is characterized by a distinctive developmental trajectory, with individuals progressing through four stages: early onset, rapid developmental regression, plateau or pseudo-stationary period, and late motor deterioration. Each stage is associated with specific clinical features and challenges.
Stage I
Early Onset (6-18 months)
The early onset stage is characterized by subtle signs of developmental delay that may be easily overlooked. Infants with Rett syndrome may exhibit:
- Delayed or stagnated head growth (microcephaly)
- Reduced interest in play and social interaction
- Hypotonia (decreased muscle tone)
- Mildly delayed motor development
- Subtle hand stereotypies (repetitive movements)
During this stage, infants may still achieve certain milestones such as sitting, crawling, and even walking, although these skills may emerge later than expected or be less refined compared to typically developing infants.
Stage II
Rapid Developmental Regression (1-4 years)
The rapid developmental regression stage is the most distinctive and diagnostically significant phase of Rett syndrome. This stage is characterized by a profound loss of previously acquired skills and the emergence of characteristic features:
- Loss of purposeful hand skills and the development of repetitive, stereotypic hand movements such as hand wringing, clapping, tapping, and mouthing
- Regression or loss of spoken language, including babbling and any acquired words
- Impaired social interaction and communication, with features reminiscent of autism spectrum disorder
- Gait abnormalities, including an unsteady, wide-based, or stiff-legged gait, or loss of the ability to walk in some cases
- Breathing irregularities, such as hyperventilation, breath-holding, or air swallowing, particularly during wakefulness
- Seizures, which may emerge during this stage or later in the course of the disorder
The rapid regression stage is often the most distressing for families, as the child's previously acquired skills and engagement with the world seem to slip away. This stage typically lasts for several weeks to months, after which the child enters a period of apparent stabilization.
Stage III
Plateau Phrase Following the regression stage, children with Rett syndrome enter a plateau or pseudo-stationary period, characterized by a relative stabilization of symptoms and potential improvements in certain areas:
- Stabilization or slight improvement in hand skills, with some individuals learning to use simple hand gestures or manipulate objects
- Persistent motor difficulties, including apraxia (difficulty with purposeful movements), ataxia (uncoordinated movements), and dystonia (abnormal muscle tone)
- Improved eye contact and non-verbal communication, with many individuals developing an "intense eye gaze" for expressing needs and preferences
- Ongoing seizures, which may be more prominent during this stage and require medical management
- Development of scoliosis (curvature of the spine) in many individuals, often requiring orthopedic intervention
Despite the term "stationary," this stage is not truly static, as individuals with Rett syndrome continue to face significant challenges and may experience ongoing changes in their symptoms and abilities. However, the plateau stage often provides an opportunity for individuals to learn new skills and engage with their environment, given appropriate support and interventions.
Stage IV
Motor Deterioration Phase The late motor deterioration stage is characterized by a gradual decline in motor function and increased vulnerability to medical complications:
- Reduced mobility, with many individuals becoming wheelchair-dependent or requiring significant assistance with walking
- Increased muscle weakness, spasticity, and rigidity, contributing to contractures and deformities
- Progression of scoliosis, often requiring surgical intervention to maintain posture and prevent respiratory complications
- Decreased hand skills and increased stereotypic movements, although some individuals may retain the ability to perform simple hand gestures
- Potential improvement in communication and social interaction, with some individuals showing increased responsiveness and emotional connection
Assistive Technology and AAC Interventions
Assistive technology and augmentative and alternative communication (AAC) interventions are crucial components of the comprehensive care approach for individuals with Rett syndrome. These interventions aim to support communication, enhance independence, and improve overall quality of life by addressing the unique challenges faced by individuals with Rett syndrome, particularly in the areas of speech, language, and motor function.

Communication Devices
The loss of spoken language is one of the core features of Rett syndrome, making AAC devices essential tools for enabling individuals to express their needs, preferences, and ideas. The choice of AAC device depends on the individual's unique abilities, challenges, and communication goals, and may include:
- Eye-gaze technology: Many individuals with Rett syndrome retain the ability to control their eye movements, making eye-gaze systems a highly effective means of communication. These systems allow users to select symbols, words, or phrases on a screen by looking at them, enabling communication, computer access, and environmental control.
- Communication boards: Low-tech communication boards displaying pictures, symbols, or words can be used by individuals who have some hand function or can indicate their choices through eye gaze or other body movements. High-tech communication boards with dynamic displays and speech output can provide a wider range of vocabulary and customization options.
- Tablets with specialized software: Tablet devices equipped with AAC apps can offer a portable and versatile communication solution for individuals with Rett syndrome. These apps can be customized to the user's specific needs, providing access to a wide range of vocabulary, symbols, and communication functions.
- Switch-activated communication tools: For individuals with limited hand function, switch-activated devices can be used to access pre-recorded messages or scan through communication options. Switches can be adapted to respond to various body movements, such as head turns, arm movements, or even slight muscle contractions.
The selection and implementation of AAC devices should be guided by a comprehensive assessment of the individual's communication needs, cognitive abilities, motor skills, and sensory functions. The assessment process should involve a multidisciplinary team, including speech-language pathologists, occupational therapists, and assistive technology specialists, working in close collaboration with the individual, their family, and educational staff.
Mobility Aids
Motor impairments, including apraxia, ataxia, and muscle weakness, are common in Rett syndrome and can significantly impact an individual's ability to move independently and participate in daily activities. Assistive technology for mobility can help individuals maintain or improve their functional abilities and prevent secondary complications associated with immobility. Mobility aids may include:
- Wheelchairs: Manual or power wheelchairs can provide independent mobility for individuals who are unable to walk or have difficulty walking safely. Wheelchair seating systems should be customized to provide best support, comfort, and pressure relief, taking into account the individual's posture, muscle tone, and risk of developing scoliosis or other orthopedic complications.
- Walkers: For individuals who have some walking ability but require support for balance and stability, walkers such as gait trainers or posterior walkers can be used. These devices provide a stable base of support and can be adjusted to accommodate the individual's height, posture, and level of assistance needed.
- Standing frames: Regular standing can help improve bone density, respiratory function, and gastrointestinal motility, as well as provide opportunities for social interaction and participation in activities. Standing frames support an upright posture and can be used for individuals who are unable to stand independently or require assistance to maintain a standing position.
The selection of mobility aids should be based on a comprehensive assessment of the individual's motor abilities, postural needs, and functional goals, conducted by a team of physical therapists, occupational therapists, and assistive technology specialists. Regular monitoring and adjustments are necessary to ensure that the mobility aids continue to meet the individual's changing needs and promote best function and participation.
Access Modalities
Assistive Technology Interventions
The success of assistive technology interventions for individuals with Rett syndrome largely depends on the selection of appropriate access modalities that accommodate the individual's motor abilities and cognitive skills. The most common access modalities used by individuals with Rett syndrome include:
- Eye-gaze technology: Eye-gaze systems are often the most effective access method for individuals with Rett syndrome, as eye movement control is relatively preserved compared to other motor functions. Eye-gaze devices use cameras to track the user's eye movements and translate them into cursor movements or selections on a screen. With proper calibration and training, individuals can use eye-gaze technology to access communication software, computer applications, and environmental control systems.
- Switches: Adaptive switches can be used to access assistive technology devices by detecting intentional movements or muscle contractions. Switches come in various shapes, sizes, and activation methods (e.g., pressure, proximity, or muscle movement) and can be positioned to capitalize on the individual's most reliable and consistent motor skills. Individuals can use switches to activate pre-programmed messages, scan through communication options, or control devices in their environment.
- Touch access: For individuals with some hand function, touch screens or large button interfaces may be appropriate for accessing communication devices or other assistive technologies. Touch access can be enhanced by using strategies such as key guards, which provide physical boundaries around buttons to prevent accidental selections, or by adjusting the sensitivity and response time of the touch screen.
- Head-pointing devices: Head-pointing systems use infrared cameras or reflective markers to track the user's head movements and translate them into cursor movements on a screen. These devices can be used to access communication software, computer applications, or environmental control systems for individuals who have reliable head control but limited hand function.
The process of selecting and implementing access modalities for individuals with Rett syndrome should involve a collaborative assessment by a multidisciplinary team, including occupational therapists, speech-language pathologists, and assistive technology specialists. The team should consider the individual's current motor abilities, potential for skill development, and the demands of the specific assistive technology devices being used. Ongoing training and support for the individual, family members, and educational staff are essential to ensure the successful integration of access modalities into daily life and to promote the individual's maximum participation and independence.
Environmental Control Units (ECUs)
Environmental control units (ECUs) are assistive technology devices that enable individuals with Rett syndrome to control various aspects of their environment, such as lights, appliances, doors, and communication devices, using alternative access methods like switches or eye-gaze systems. By providing a means to interact with their surroundings, ECUs can enhance independence, self-determination, and quality of life for individuals with significant motor impairments.
ECUs can be programmed to control a wide range of devices and functions, depending on the individual's needs and preferences:
- Lights: Turning lights on and off, adjusting brightness, or changing colors.
- Entertainment devices: Operating televisions, music players, or other media devices.
- Doors and windows*: Opening and closing doors, curtains, or blinds.
- Climate control: Adjusting room temperature or fan settings.
- Communication devices: Activating pre-programmed messages or initiating communication functions on AAC devices.
- Call systems: Alerting caregivers or family members when assistance is needed.
The integration of ECUs into the daily lives of individuals with Rett syndrome requires careful assessment, customization, and training to ensure that the system is tailored to the individual's unique abilities, needs, and environment. Occupational therapists and assistive technology specialists play a key role in evaluating the individual's control and access options, selecting appropriate ECU components, and providing training to the individual, family members, and caregivers.
Clinical Recommendations

Recommendations for Speech-Language Pathologists
- Comprehensive Assessment:
- Conduct a thorough assessment of communication skills, including receptive and expressive language abilities, nonverbal communication, and AAC competencies.
- Evaluate oral-motor skills, including the ability to produce vocalizations, control oral secretions, and manage food and drink textures safely.
- Assess auditory processing skills and identify any sensory factors that may impact communication performance.
- Expressive Language Intervention:
- Target the development of functional communication skills that are relevant to daily life and promote independence and self-advocacy.
- Implement a core vocabulary approach, focusing on high-frequency words that can be used across various contexts and communication partners.
- Incorporate a range of AAC modalities, such as eye gaze access, switch activation, and low-tech communication boards, to provide multiple opportunities for expressive output.
- Teach how to combine symbols to express more complex ideas, feelings, and social messages.
- Receptive Language Intervention:
- Use visual supports, such as pictures, objects, and written words, to enhance understanding of spoken language and to provide a permanent reference for learning.
- Break down complex instructions and information into smaller, more manageable chunks, using simple language and repetition to reinforce comprehension.
- Incorporate interests and motivators into receptive language activities to increase engagement and retention of new concepts.
- Teach how to follow increasingly complex directions and to respond to a variety of question types (e.g., yes/no, what, where, who) using AAC systems.
- Social Communication Intervention:
- Target the development of social communication skills, such as initiating and maintaining conversations, taking turns, and expressing emotions and empathy.
- Use role-play, social stories, and video modeling to teach and practice social communication behaviors in a structured and supportive environment.
- Incorporate peer-mediated interventions, such as cooperative learning groups or buddy systems, to provide opportunities for authentic social interactions with peers.
- Coach communication partners, including family members, teachers, and support staff, on strategies to facilitate successful social exchanges and to create an inclusive communication environment.
- AAC System Optimization:
- Continuously monitor and update AAC systems to ensure they meet evolving communication needs and reflect expanding vocabulary and language skills.
- Collaborate with occupational therapists and assistive technology specialists to optimize physical access to AAC devices, considering factors such as positioning, switch placement, and visual adaptations.
- Provide ongoing training and support to communication partners to ensure they are confident and competent in using and maintaining AAC systems.
- Generalization and Maintenance:
- Implement strategies to promote the generalization of communication skills across various settings, people, and activities, such as using a consistent vocabulary and prompting hierarchy across environments.
- Develop home programs that empower family members to support communication development through daily routines, play activities, and shared reading experiences.
- Establish systems for regular monitoring and data collection to track progress, identify areas for further intervention, and celebrate successes.
- Foster a culture of open communication and collaboration among team members to ensure that communication needs are consistently met and that intervention strategies are continually refined based on response and progress.
Recommendations for Applied Behavior Analysis (ABA)
- Functional Behavior Assessment (FBA):
- Conduct FBAs to identify the antecedents, consequences, and functions of challenging behaviors, such as self-injury, aggression, or non-compliance.
- Use a variety of data collection methods, including direct observation, caregiver interviews, and environmental assessments, to gather comprehensive information about behavior patterns.
- Develop hypotheses about the underlying reasons for challenging behaviors, considering factors such as sensory needs, communication difficulties, or skill deficits.
- Positive Behavior Support Plans (PBSPs):
- Based on FBA results, develop PBSPs that outline proactive strategies, replacement behavior teaching, and response protocols to address challenging behaviors.
- Incorporate antecedent modifications, such as environmental accommodations, visual supports, or sensory strategies, to prevent or minimize the occurrence of challenging behaviors.
- Identify socially appropriate replacement behaviors that serve the same function as the challenging behavior and teach these skills using systematic instruction and reinforcement.
- Develop consistent and compassionate response protocols for managing challenging behaviors when they occur, focusing on de-escalation, redirection, and positive reinforcement of alternative behaviors.
- Skill Acquisition Programs:
- Design and implement individualized skill acquisition programs that target adaptive behavior, self-help, and independence goals.
- Use task analysis to break down complex skills into smaller, more manageable steps and teach each step using prompting, shaping, and chaining procedures.
- Incorporate visual supports, such as picture schedules, step-by-step guides, or video models, to enhance understanding and independence in completing tasks.
- Utilize positive reinforcement strategies, such as token economies, choice boards, or differential reinforcement, to increase motivation and engagement in learning new skills.
- Data-Driven Decision Making:
- Establish systems for ongoing data collection and analysis to monitor progress, evaluate the effectiveness of interventions, and inform data-driven decision making.
- Use a variety of data collection methods, such as frequency counts, duration recording, or time sampling, to capture accurate and reliable information about behaviors and skill acquisition.
- Regularly review and analyze data with educational teams to identify trends, patterns, and areas for modification or intensification of intervention strategies.
- Communicate data findings to family members and other stakeholders in a clear and accessible manner, using visual displays and progress reports to facilitate understanding and collaboration.
- Caregiver Training and Support:
- Provide ongoing training and support to family members and caregivers to ensure the consistent implementation of ABA strategies across all settings.
- Teach caregivers how to use positive reinforcement, prompting, and error correction procedures to support skill development and behavior management.
- Coach caregivers on strategies for promoting generalization and maintenance of skills, such as incorporating learning opportunities into daily routines and natural environments.
- Offer emotional support and resources to help caregivers manage the unique challenges and stressors associated with caring for a child with complex needs.
- Collaboration and Coordination:
- Work closely with other members of educational and therapeutic teams, including speech-language pathologists, occupational therapists, and special educators, to ensure a coordinated and comprehensive approach to intervention.
- Participate in regular team meetings and case conferences to share information, discuss progress, and collaboratively problem-solve any challenges or barriers to success.
- Provide consultation and training to school staff and community providers to build capacity and ensure the consistent implementation of ABA principles and strategies across all relevant settings.
- Advocate for the needs and rights of individuals with complex communication needs within the educational system and community, promoting a culture of inclusion, respect, and high expectations.
Recommendations for Occupational Therapists
Fine Motor Skills:
- Provide opportunities to practice reaching, grasping, and releasing objects to maintain and improve hand function.
- Incorporate eye-hand coordination activities to support the development of purposeful hand movements.
- Collaborate with SLPs to integrate fine motor goals with AAC use, such as activating switches or pointing to symbols.
Sensory Processing:
- Conduct comprehensive sensory assessments to identify sensory processing patterns and their impact on functioning.
- Develop personalized sensory diets that incorporate calming and alerting strategies to help regulate arousal levels throughout the day.
- Provide sensory-based interventions, such as deep pressure input or tactile stimulation, to reduce hand stereotypies and promote focused engagement.
Assistive Technology:
- Assess positioning needs and recommend adaptive equipment, such as seating systems or standers, to optimize alignment and stability for AAC access.
- Collaborate with SLPs to identify appropriate switch placement and mounting options for AAC devices.
- Train staff and family members on the proper use and positioning of assistive technology to ensure consistent implementation across environments.
Activities of Daily Living (ADLs):
- Assess current levels of independence in ADLs, such as feeding, dressing, and hygiene routines.
- Develop strategies to promote participation and independence in ADLs, such as adapted utensils, dressing aids, or visual supports.
- Provide training to staff and family members on techniques to support engagement in ADLs while respecting autonomy and preferences.
Environmental Modifications:
- Assess physical environments to identify barriers to participation and independence.
- Recommend modifications, such as accessible furniture, lighting adjustments, or sound-absorbing materials, to create supportive and inclusive environments.
- Collaborate with teams to ensure that environmental modifications are consistent across school, home, and community settings.
Recommendations for All Staff and Caregivers
AAC Implementation:
- Ensure AAC devices are within line of sight at all times, maximizing opportunities for communication.
- Model the use of AAC devices in all interactions, even when not actively being used, providing consistent examples of AAC use.
- Provide ample wait time (at least 10 seconds) after asking questions or presenting choices, allowing time to process and respond.
Communication Opportunities:
- Create structured communication opportunities throughout the day (e.g., choosing activities, expressing needs), promoting regular AAC practice.
- Use visual schedules to support transitions and provide opportunities to indicate preferences, enhancing predictability and choice-making.
Sensory Considerations:
- Implement sensory diets as prescribed by occupational therapists, addressing sensory processing needs that impact communication.
- Offer hand fidgets or sensory activities before communication sessions to reduce hand stereotypies, optimizing focus and access.
Positive Behavior Support:
- Use communication profiles to interpret potential meanings behind challenging behaviors, guiding appropriate responses.
- Redirect to AAC devices when frustration or anxiety appears, promoting communication as a coping strategy.
Consistency in Approach:
- Use agreed-upon hand-under-hand guidance techniques when supporting AAC use, providing consistent motor support.
- Maintain a calm, positive demeanor even when communication attempts are unsuccessful, fostering supportive environments.
Data Collection:
- Record daily communication successes and challenges in provided logs, enabling data-driven decision making.
- Note any new gestures, vocalizations, or AAC symbols used, capturing emerging skills and progress.
Environmental Adaptations:
- Ensure proper positioning for best AAC access (consult with OTs for specific guidelines), maximizing physical access to devices.
- Minimize visual and auditory distractions during communication sessions, creating focused learning environments.
Team Collaboration:
- Attend monthly team meetings to discuss progress and challenges, ensuring a coordinated approach to intervention.
- Share observations and insights with speech-language pathologists and behavior analysts, contributing to a comprehensive understanding of needs.
Family Involvement:
- Support family visits by modeling appropriate communication strategies, promoting generalization of skills to home environments.
- Provide families with updates on communication progress and any new strategies being implemented, fostering continuity of care.
Continuing Education:
- Participate in quarterly in-service trainings on Rett syndrome and AAC, staying current on best practices and research.
- Review and practice new communication strategies introduced by SLPs, ensuring fidelity of implementation.
Recommendations for Special Educators
Individualized Education Plans (IEPs):
- Collaborate with multidisciplinary teams to develop IEPs that address unique strengths, needs, and learning styles.
- Ensure that communication goals are embedded throughout educational programs, with a focus on generalizing AAC use across academic and social contexts.
- Regularly review and update IEPs based on progress, assessment data, and input from team members and families.
Curriculum Adaptations:
- Modify instructional materials and activities to ensure accessibility and meaningful participation.
- Incorporate interests and preferences into lesson plans to increase motivation and engagement.
- Use multi-sensory approaches to instruction, integrating visual, auditory, and tactile elements to support learning.
Inclusive Practices:
- Foster inclusive classroom environments that value diversity and promote acceptance of individual differences.
- Provide opportunities for structured social interactions with peers, using facilitation strategies and visual supports as needed.
- Collaborate with support staff and related service providers to ensure that accommodations and modifications are consistently implemented across all educational settings.
Transition Planning:
- Begin transition planning early to identify long-term goals and support needs beyond the educational setting.
- Collaborate with families, adult service providers, and community agencies to develop comprehensive transition plans that address areas such as employment, independent living, and community participation.
- Provide opportunities to explore interests, strengths, and preferences through community-based experiences and vocational activities.
Professional Development:
- Engage in ongoing professional development opportunities to stay current on best practices in educating students with Rett syndrome and other complex communication needs.
- Collaborate with related service providers and medical professionals to gain a holistic understanding of needs and to ensure consistency in intervention approaches.
- Share knowledge and expertise with colleagues to build capacity and foster a culture of continuous improvement in supporting students with significant disabilities.
Summing Up
Rett syndrome is a complex neurodevelopmental disorder that presents significant challenges for affected individuals and their families. However, with a comprehensive, multidisciplinary approach to care, individuals with Rett syndrome can achieve meaningful improvements in their quality of life and participation in daily activities.
The management of Rett syndrome involves a wide range of interventions, including assistive technology and AAC devices to support communication, mobility aids to enhance functional mobility, and therapeutic interventions to address motor, sensory, and cognitive impairments. Medical management is also critical for addressing the various health issues associated with Rett syndrome, such as seizures, gastrointestinal problems, and orthopedic complications.
Psychosocial support is an essential component of care, helping families cope with the emotional and practical challenges of caring for a child with Rett syndrome and promoting the individual's social and emotional well-being.
Ongoing research into the genetic basis of Rett syndrome, the development of targeted therapies, and the refinement of educational and rehabilitative approaches holds promise for improving outcomes and enhancing the lives of those affected by this disorder.
Ultimately, the goal of care for individuals with Rett syndrome is to maximize their potential, minimize complications, and ensure they can participate in meaningful activities and relationships to the fullest extent possible. Through collaborative and compassionate care, healthcare professionals, educators, and families can make a profound difference in the lives of those affected by Rett syndrome, helping them to thrive despite the challenges they face.
Positioning
Comprehensive Positioning Assessments:
- Collaborate with occupational therapists (OTs) and physical therapists (PTs) to conduct comprehensive assessments of positioning needs across various activities and environments.
- Evaluate postural control, muscle tone, range of motion, and any orthopedic or neuromuscular conditions that may impact positioning.
- Assess functional abilities, such as the ability to maintain head control, reach for objects, or engage in fine motor tasks, in different positions.
- Consider personal preferences and comfort levels when determining best positioning arrangements.
Customized Seating and Positioning Equipment:
- Based on the results of positioning assessments, recommend and acquire customized seating and positioning equipment that supports unique needs.
- Consider factors such as postural alignment, pressure distribution, and adjustability when selecting seating systems, such as wheelchairs, standers, or adaptive chairs.
- Ensure that positioning equipment allows for proper hip, knee, and ankle alignment to prevent contractures and maintain joint integrity.
- Incorporate supportive features, such as lateral supports, headrests, or tilt-in-space options, to optimize comfort, stability, and function.
Therapeutic Positioning Interventions:
- Develop daily positioning schedules that include a variety of positions and activities to prevent prolonged periods of immobility and promote physical well-being.
- Incorporate therapeutic positioning techniques, such as prone positioning, side-lying, or supported sitting, to improve respiratory function, digestion, and overall comfort.
- Use positioning aids, such as wedges, rolls, or bolsters, to maintain proper alignment and support during various activities, such as play, learning, or rest.
- Collaborate with educational teams to ensure that positioning needs are met consistently across all school-based activities and transitions.
Pressure Relief and Skin Integrity:
- Implement rigorous pressure relief schedules to prevent the development of pressure sores and maintain skin integrity.
- Teach caregivers and school staff how to perform proper weight shifts, position changes, and skin checks to identify and address any areas of concern.
- Consider the use of pressure-relieving cushions, mattresses, or overlays to distribute weight evenly and reduce the risk of skin breakdown.
- Ensure that clothing and positioning equipment are free from wrinkles, seams, or other potential sources of skin irritation.
Caregiver Training and Follow-Up:
- Provide comprehensive training to family members and caregivers on proper positioning techniques, equipment use, and safety considerations.
- Develop positioning plans that outline specific strategies and schedules for use across home, school, and community settings.
- Conduct regular follow-up assessments to monitor positioning needs, make adjustments to equipment as necessary, and address any challenges or concerns.
- Foster open communication and collaboration among all members of care teams to ensure consistent implementation of positioning strategies and promote best outcomes.
Feeding and Swallowing
Comprehensive Feeding and Swallowing Assessments:
- Collaborate with speech-language pathologists (SLPs) and occupational therapists (OTs) to conduct comprehensive assessments of feeding and swallowing skills.
- Evaluate oral-motor skills, including the ability to manage food and drink textures, coordinate suck-swallow-breathe patterns, and control oral secretions.
- Assess postural stability and alignment during feeding, considering factors such as head control, trunk support, and seating arrangements.
- Identify any sensory factors that may impact feeding performance, such as taste, texture, or temperature preferences or aversions.
Mealtime Management Plans:
- Based on the results of feeding and swallowing assessments, develop individualized mealtime management plans that outline strategies for safe and efficient feeding.
- Determine the most appropriate food and drink textures, considering oral-motor skills, sensory preferences, and nutritional needs.
- Identify any adaptive equipment or utensils that may enhance independence and safety during mealtimes, such as specialized cups, straws, or adapted plates and utensils.
- Establish consistent mealtime routines that include proper positioning, pacing, and environmental modifications to minimize distractions and optimize feeding performance.
Oral-Motor Interventions:
- Implement oral-motor exercises and activities to strengthen and coordinate the muscles involved in feeding and swallowing, such as tongue lateralization, lip closure, and jaw grading.
- Use sensory stimulation techniques, such as oral massage, vibration, or thermal stimulation, to improve oral awareness and responsiveness.
- Incorporate oral-motor play activities, such as blowing bubbles, using straws, or exploring different food textures, to promote skill development and enjoyment of oral experiences.
Dysphagia Management:
- Monitor closely for signs and symptoms of dysphagia, such as coughing, choking, or respiratory distress during meals.
- Implement compensatory strategies, such as chin tuck, head turn, or multiple swallows, to reduce the risk of aspiration and improve swallowing safety.
- Collaborate with medical professionals, such as pulmonologists or gastroenterologists, to address any underlying medical conditions that may contribute to dysphagia, such as reflux or respiratory issues.
- Consider the use of thickening agents or modified food textures, as recommended by SLPs, to ensure safe and efficient swallowing.
Caregiver Training and Support:
- Provide comprehensive training to family members and caregivers on safe feeding techniques, positioning, and emergency management procedures.
- Teach caregivers how to recognize signs of distress or aspiration during meals and how to respond appropriately.
- Develop systems for tracking food and fluid intake, as well as any mealtime challenges or successes, to inform ongoing adjustments to feeding plans.
- Offer emotional support and resources to help caregivers manage the stress and anxiety associated with feeding difficulties and ensure positive and enjoyable mealtime experiences for all involved.
Care Management and Resources
The management of Rett syndrome requires a comprehensive, multidisciplinary approach that addresses the complex and evolving needs of affected individuals throughout their lifespan. The primary goals of care are to optimize functional abilities, minimize complications, and enhance the quality of life for individuals with Rett syndrome and their families.
Psychosocial Impact on Families
Receiving a Rett syndrome diagnosis can have a profound psychosocial impact on families. Parents often experience a complex range of emotions, including shock, grief, anger, and anxiety about the future. Siblings may also struggle with adjusting to the changes in family dynamics and the increased attention given to their affected sister.
Healthcare professionals should:
- Provide empathetic, non-judgmental support and validation of the family's emotional experiences.
- Offer referrals to mental health professionals, such as psychologists or counselors, who have experience working with families of children with disabilities.
- Connect families with local and online support groups, where they can share experiences, resources, and coping strategies with others who understand their unique challenges.
- Provide education about respite care services and encourage families to prioritize self-care and stress management.
- Collaborate with social workers to help families navigate complex healthcare and educational systems, access financial assistance programs, and plan for long-term care needs.
- Recognize that the psychosocial needs of families may change over time, and regularly assess for signs of caregiver burnout, depression, or marital strain.
- Celebrate the child's strengths and milestones, and help families maintain hope and a positive outlook despite the challenges they face.
By addressing the psychosocial needs of families in a holistic and proactive manner, healthcare professionals can promote resilience, empowerment, and improved quality of life for all family members affected by Rett syndrome.
IEP Goal Examples
Communication
- Using her eye gaze AAC device, [Student] will select from a field of 4 symbols to request preferred items or activities during snack and leisure times, with 80% accuracy across 3 consecutive sessions.
- When presented with a simple question (e.g., "Do you want to listen to music?"), [Student] will respond yes/no using a switch-activated voice output device, with 90% accuracy across 5 consecutive opportunities.
Assistive Technology
- [Student] will use an adapted joystick to navigate her power wheelchair independently from the classroom to the cafeteria, making no more than 2 verbal prompts, for 4 out of 5 opportunities.
- Using a head-mounted Proximity Switch, [Student] will activate a toy or simple appliance (e.g., blender, radio) to participate in cause-effect activities, with physical prompts faded to independence across 6 weeks.
Functional Skills
- With hand-under-hand assistance, [Student] will bring a pre-loaded spoon from the bowl to her mouth to complete 5 bites of preferred foods during lunchtime, for 3 consecutive days.
- During morning circle time, [Student] will use her eye gaze AAC device to select the day of the week and weather conditions from an array of 6 symbols, with 85% accuracy across 2 consecutive weeks.
Accommodations
- Provide extended time (1.5x) for [Student] to respond to questions or make selections using her AAC system.
- Use visual schedules and timers to support [Student]'s understanding of daily routines and expectations.
- Ensure that [Student]'s positioning equipment is properly adjusted and all necessary AAC components are charged and accessible prior to each lesson or activity.
Transition Planning
As students with Rett syndrome approach adulthood, comprehensive transition planning is essential to ensure a smooth progression from school-based services to adult supports. Key areas to address in the transition process include:
Post-Secondary Education
- Research college programs that offer specialized support for students with intellectual and developmental disabilities, such as inclusive higher education or vocational training programs.
- Identify scholarship opportunities and funding sources, such as the Federal Pell Grant or state-specific financial aid programs for students with disabilities.
Employment
- Conduct vocational assessments to identify the student's strengths, interests, and potential job matches.
- Provide job shadowing, internship, or volunteer experiences to help the student build relevant skills and explore different career paths.
- Connect with local disability employment agencies, such as Vocational Rehabilitation or Workforce Development Centers, to access job coaching, assistive technology, and placement services.
Independent Living
- Assess the student's functional living skills and identify areas where additional training or support may be needed, such as money management, meal preparation, or self-care routines.
- Explore housing options, such as supported living arrangements, group homes, or assistive technology-equipped apartments, based on the student's level of independence and care needs.
- Provide training in self-advocacy skills, such as communicating accommodation needs, managing healthcare appointments, and participating in person-centered planning meetings.
Community Participation
- Identify local recreational programs, adaptive sports leagues, or social clubs that align with the student's interests and abilities.
- Teach skills for accessing community resources, such as using public transportation, shopping, or attending religious services, with appropriate accommodations and support.
- Foster connections with disability advocacy organizations and peer support groups to promote a sense of belonging and empowerment.
Long-Term Care
- Assist families in applying for adult disability services, such as Medicaid Waivers or Supplemental Security Income (SSI), to ensure continuity of care and financial support.
- Develop a comprehensive care plan that outlines the student's medical, therapeutic, and daily living needs, and identifies responsible parties for overseeing each aspect of care.
- Explore legal options for guardianship, power of attorney, or supported decision-making, based on the student's cognitive abilities and support needs.
By initiating the transition planning process early (ideally by age 14) and collaborating closely with the student, family, and adult service providers, educational teams can help ensure that students with Rett syndrome have the skills, supports, and opportunities necessary to lead fulfilling, self-determined lives as adults. Regular monitoring and adjustments to the transition plan will be necessary as the student's needs, preferences, and circumstances evolve over time.
Organizations and Support
Wrapping Up
The goal of this guide was to synthesize a wide body of evidence-based practices and research on Rett syndrome to provide a thorough overview of the disorder's clinical features, diagnostic criteria, and management strategies across key domains of functioning. While the information presented aligns with current standards of care and scientific consensus, it is not intended to replace the professional judgment of practitioners in the fields of medicine, allied health, education, or behavior analysis.
Success in supporting individuals with Rett syndrome relies on a strong commitment to person-centered care, ongoing collaboration among multidisciplinary team members, and a shared vision of each individual's unique potential for growth and achievement. As individuals with Rett syndrome continue to develop and face new challenges throughout their lifespan, it is essential that their care plans remain flexible, responsive, and grounded in a holistic approach that values their autonomy, dignity, and distinctive contributions.
A critical component of effective management is the coordination and integration of services across multiple disciplines, including medicine, speech-language pathology, occupational therapy, physical therapy, psychology, applied behavior analysis, and special education. By fostering open communication, shared decision-making, and a unified approach to intervention, care teams can ensure that the complex needs of individuals with Rett syndrome are met consistently and comprehensively across all environments.
Furthermore, the active involvement and empowerment of families and caregivers are vital to promoting the generalization and maintenance of skills, as well as ensuring a high quality of life for both the affected individual and their support system. Providing ongoing education, training, and resources to families and caregivers enables them to effectively advocate for their loved one's rights, implement therapeutic strategies at home, and navigate the unique challenges and joys of caring for someone with Rett syndrome.
As individuals with Rett syndrome progress through different life stages, it is crucial to engage in proactive planning to address their evolving needs and facilitate smooth transitions between services. This process should involve exploring opportunities for continued learning, skill development, community participation, and maximizing independence to the greatest extent possible, tailored to each individual's strengths, preferences, and goals. By anticipating and preparing for future challenges and milestones, care teams can help ensure best outcomes and quality of life across the lifespan.
Ultimately, by embracing the inherent worth and potential of every individual with Rett syndrome, addressing their multifaceted needs, and advocating for their fundamental human rights, we can create a more inclusive, supportive, and empowering society that enables them to thrive and lead fulfilling lives. This guide serves as a comprehensive resource and roadmap for that journey, informing evidence-based practices and guiding care teams towards a brighter, more hopeful future for all those impacted by Rett syndrome. With unwavering dedication, scientific rigor, and a profound respect for the resilience and spirit of these remarkable individuals, we can continue to advance our understanding and transform the landscape of care for Rett syndrome, one impossible thing at a time.
References for This Page
- Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics, 23(2), 185-188. https://doi.org/10.1038/13810
- Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi-Buisson, N., Leonard, H., Bailey, M. E. S., Schanen, N. C., Zappella, M., Renieri, A., Huppke, P., Percy, A. K., & RettSearch Consortium. (2010). Rett syndrome: Revised diagnostic criteria and nomenclature. Annals of Neurology, 68(6), 944-950. https://doi.org/10.1002/ana.22124
- Hagberg, B. (2002). Clinical manifestations and stages of Rett syndrome. Mental Retardation and Developmental Disabilities Research Reviews, 8(2), 61-65. https://doi.org/10.1002/mrdd.10020
- Kaufmann, W. E., Tierney, E., Rohde, C. A., Suarez-Pedraza, M. C., Clarke, M. A., Salorio, C. F., Bibat, G., Bukelis, I., Naram, D., Lanham, D. C., & Naidu, S. (2012). Social impairments in Rett syndrome: Characteristics and relationship with clinical severity. Journal of Intellectual Disability Research, 56(3), 233-247. https://doi.org/10.1111/j.1365-2788.2011.01404.x
- Cianfaglione, R., Clarke, A., Kerr, M., Hastings, R. P., Oliver, C., & Felce, D. (2015). A national survey of Rett syndrome: Behavioural characteristics. Journal of Neurodevelopmental Disorders, 7(1), 11. https://doi.org/10.1186/s11689-015-9104-y
- Townend, G. S., Marschik, P. B., Smeets, E., van de Berg, R., van den Berg, M., & Curfs, L. M. G. (2016). Eye gaze technology as a form of augmentative and alternative communication for individuals with Rett syndrome: Experiences of families in the Netherlands. Journal of Developmental and Physical Disabilities, 28(1), 101-112. https://doi.org/10.1007/s10882-015-9455-z
- Lotan, M., & Zysman, L. (2011). The digestive system and nutritional considerations in Rett syndrome. In A. Lotan & J. Merrick (Eds.), Rett Syndrome (pp. 113-130). Nova Science Publishers.
- Leonard, H., Cobb, S., & Downs, J. (2017). Clinical and biological progress over 50 years in Rett syndrome. Nature Reviews Neurology, 13(1), 37-51. https://doi.org/10.1038/nrneurol.2016.186
- Fabio, R. A., Billeci, L., Crifaci, G., Troise, E., Tortorella, G., & Pioggia, G. (2016). Cognitive training modifies frequency EEG bands and neuropsychological measures in Rett syndrome. Research in Developmental Disabilities, 53-54, 73-85. https://doi.org/10.1016/j.ridd.2016.01.009
- Motil, K. J., Caeg, E., Barrish, J. O., Geerts, S., Lane, J. B., Percy, A. K., Annese, F., McNair, L., Skinner, S. A., Lee, H.-S., Neul, J. L., & Glaze, D. G. (2012). Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. Journal of Pediatric Gastroenterology and Nutrition, 55(3), 292-298. https://doi.org/10.1097/MPG.0b013e31824b6159
- Lotan, M., & Hanks, S. (2006). Physical therapy intervention for individuals with Rett syndrome. TheScientificWorldJournal, 6, 1314-1338. https://doi.org/10.1100/tsw.2006.187
- Djukic, A., & McDermott, M. V. (2012). Social preferences in Rett syndrome. Pediatric Neurology, 46(4), 240-242. https://doi.org/10.1016/j.pediatrneurol.2012.01.011
- Sigafoos, J., Green, V. A., Schlosser, R., O'Reilly, M. F., Lancioni, G. E., Rispoli, M., & Lang, R. (2009). Communication intervention in Rett syndrome: A systematic review. Research in Autism Spectrum Disorders, 3(2), 304-318. https://doi.org/10.1016/j.rasd.2008.09.006
- Simacek, J., Reichle, J., & McComas, J. J. (2016). Communication intervention to teach requesting through aided AAC for two learners with Rett syndrome. Journal of Developmental and Physical Disabilities, 28(1), 59-81. https://doi.org/10.1007/s10882-015-9423-7
- Cuddapah, V. A., Pillai, R. B., Shekar, K. V., Lane, J. B., Motil, K. J., Skinner, S. A., Tarquinio, D. C., Glaze, D. G., McGwin, G., Kaufmann, W. E., Percy, A. K., Neul, J. L., & Olsen, M. L. (2014). Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. Journal of Medical Genetics, 51(3), 152-158. https://doi.org/10.1136/jmedgenet-2013-102113
- Killian, J. T., Lane, J. B., Lee, H.-S., Skinner, S. A., Kaufmann, W. E., Glaze, D. G., Neul, J. L., & Percy, A. K. (2016). Scoliosis in Rett syndrome: Progression, comorbidities, and predictors. Pediatric Neurology, 70, 20-25. https://doi.org/10.1016/j.pediatrneurol.2016.12.020
- Downs, J., Torode, I., Wong, K., Ellaway, C., Elliott, E. J., Christodoulou, J., Jacoby, P., Thomson, M. R., Izatt, M. T., Askin, G. N., McPhee, B. I., Bridge, C., Cundy, P., & Leonard, H. (2016). The natural history of scoliosis in females with Rett syndrome. Spine, 41(10), 856-863. https://doi.org/10.1097/BRS.0000000000001399
- Tarquinio, D. C., Motil, K. J., Hou, W., Lee, H.-S., Glaze, D. G., Skinner, S. A., Neul, J. L., Annese, F., McNair, L., Barrish, J. O., Geerts, S. P., Lane, J. B., & Percy, A. K. (2012). Growth failure and outcome in Rett syndrome: Specific growth references. Neurology, 79(16), 1653-1661. https://doi.org/10.1212/WNL.0b013e31826e9a70
- Downs, J., Rodger, J., Li, C., Tan, X., Hu, N., Wong, K., de Klerk, N., & Leonard, H. (2018). Environmental enrichment intervention for Rett syndrome: An individually randomised stepped wedge trial. Orphanet Journal of Rare Diseases, 13(1), 3. https://doi.org/10.1186/s13023-017-0752-8
- Dy, M. E., Waugh, J. L., Sharma, N., O'Leary, H., Kapur, K., D'Gama, A. M., Sahin, M., Urion, D. K., & Kaufmann, W. E. (2017). Defining hand stereotypies in Rett syndrome: A movement disorders perspective. Pediatric Neurology, 75, 91-95. https://doi.org/10.1016/j.pediatrneurol.2017.07.001
- Stallworth, J. L., Dy, M. E., Buchanan, C. B., Chen, C. F., Glaze, D. G., Lane, J. B., Li, X., Lieberman, D. N., Massey, S. L., McCormick, F., Neul, J. L., Percy, A. K., & Skinner, S. A. (2019). Hand stereotypies: Lessons from the Rett Syndrome Natural History Study. Neurology, 92(22), e2594-e2603. https://doi.org/10.1212/WNL.0000000000007560
- Tarquinio, D. C., Hou, W., Neul, J. L., Lane, J. B., Barnes, K. V., O'Leary, H. M., Bruck, N. M., Kaufmann, W. E., Motil, K. J., Glaze, D. G., Skinner, S. A., Annese, F., Baggett, L., Barrish, J. O., Geerts, S. P., Percy, A. K., & The Age of Diagnosis in Rett Syndrome Study Group. (2015). Age of diagnosis in Rett syndrome: Patterns of recognition among diagnosticians and risk factors for late diagnosis. Pediatric Neurology, 52(6), 585-591.e2. https://doi.org/10.1016/j.pediatrneurol.2015.02.007
- Percy, A. K., Neul, J. L., Glaze, D. G., Motil, K. J., Skinner, S. A., Khwaja, O., Lee, H.-S., Lane, J. B., Barrish, J. O., Annese, F., McNair, L., Graham, J., & Barnes, K. (2010). Rett syndrome diagnostic criteria: Lessons from the Natural History Study. Annals of Neurology, 68(6), 951-955. https://doi.org/10.1002/ana.22154
- Einspieler, C., Freilinger, M., & Marschik, P. B. (2016). Behavioural biomarkers of typical Rett syndrome: Moving towards early identification. Wiener Medizinische Wochenschrift, 166(11-12), 333-337. https://doi.org/10.1007/s10354-016-0498-2
- Marschik, P. B., Kaufmann, W. E., Sigafoos, J., Wolin, T., Zhang, D., Bartl-Pokorny, K. D., Pini, G., Zappella, M., Tager-Flusberg, H., Einspieler, C., & Johnston, M. V. (2013). Changing the perspective on early development of Rett syndrome. Research in Developmental Disabilities, 34(4), 1236-1239. https://doi.org/10.1016/j.ridd.2013.01.014
- Townend, G. S., Bartolotta, T. E., Urbanowicz, A., Wandin, H., & Curfs, L. M. G. (2020). Developing a model of best practice for professionals working with people with Rett syndrome. Advances in Neurodevelopmental Disorders, 4(2), 145-157. https://doi.org/10.1007/s41252-020-00152-z
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Rett Syndrome?
What does Rett Syndrome do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.