Introduction
Spinal Muscular Atrophy (SMA) is a genetic neuromuscular disorder caused by deletions or mutations in the SMN1 gene, leading to degeneration of motor neurons and muscle weakness. The disease varies in severity, with four main types classified by age of onset and motor function. Diagnosis involves clinical evaluation, genetic testing, and electromyography. Management requires a multidisciplinary approach, including pharmacological treatments, respiratory and nutritional support, physical and occupational therapy, speech and language therapy, orthopedic interventions, and lifestyle modifications. Assistive technology, such as wheelchairs, speech generating devices, and adaptive utensils, can support independence and quality of life. Ongoing research aims to improve understanding and treatment of SMA.
Spinal Muscular Atrophy (SMA) Handout
Demographic Information
- Incidence: Approximately 1 in 6,000 to 1 in 10,000 live births
- Prevalence: About 1 in 10,000 people
- Gender: Affects both genders equally
- Onset Age: Varies by type; can be present at birth or develop later in childhood or adulthood
Coding
- ICD-11: 8C70
- ICD-10-CM: G12.9
- OMIM: 253300
- UMLS: C0026650
- MeSH: D020346
- GARD: 7736
Quick Reference
| Quick Facts | Details |
|---|---|
| Incidence | 1 in 6,000 to 1 in 10,000 live births |
| Prevalence | Approximately 1 in 10,000 people |
| Gender Distribution | Affects both genders equally (autosomal recessive) |
| Primary Age of Onset | Varies by type: Type 1 (birth-6 months), Type 2 (6-18 months), Type 3 (after 18 months), Type 4 (adulthood) |
| AT/AAC Requirements | High - Varies by type; Type 1 & 2 require significant AT for mobility, communication, and respiratory support |
Medical Features and Pathophysiology
Etiology
Spinal Muscular Atrophy (SMA) is caused by deletions or mutations in the survival motor neuron 1 (SMN1) gene. This gene is crucial for the production of the SMN protein, which is essential for the survival of motor neurons. The complete loss of SMN protein is embryonically lethal, whereas reduced levels result in the selective death of motor neurons, leading to muscle paralysis.
Pathology
SMA is characterized by the degeneration of motor neurons in the spinal cord and brainstem, leading to progressive muscle weakness and atrophy. The severity of the disease varies depending on the type of SMA, which is classified based on the age of onset and the highest motor function achieved.
Symptoms
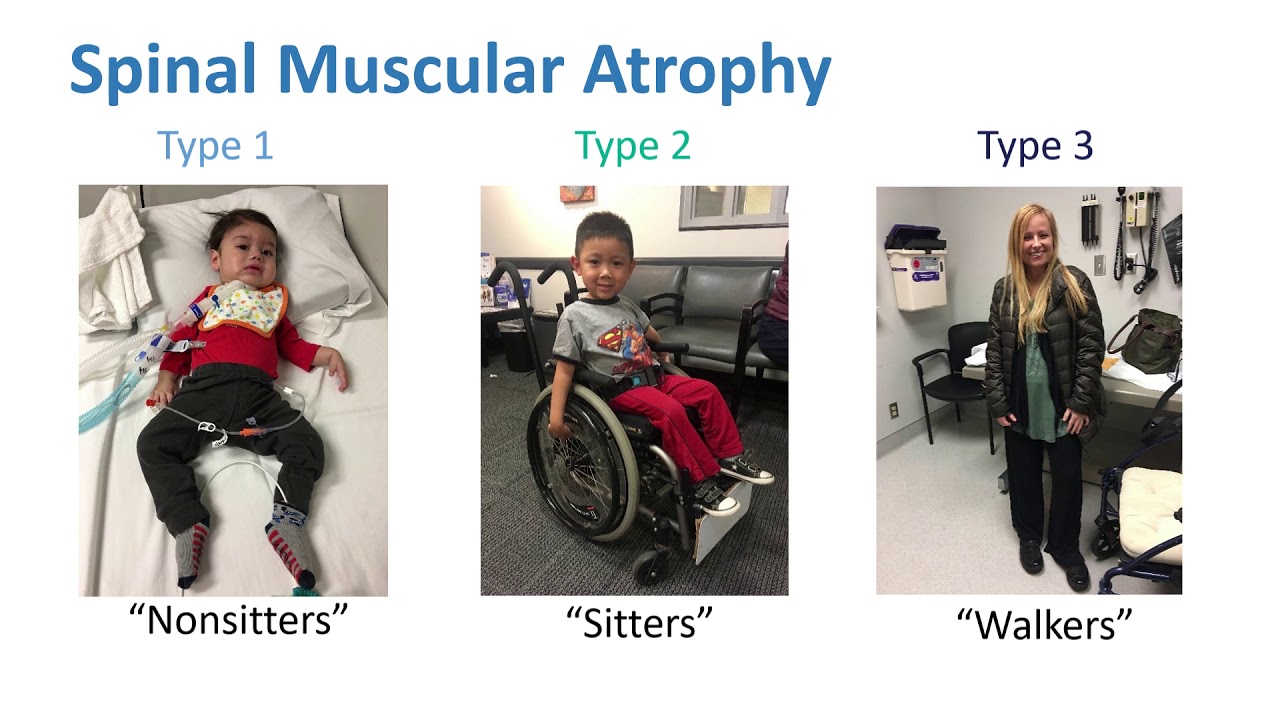
- Type 1 (Infantile Onset):
- Severe muscle weakness and hypotonia (floppy baby syndrome)
- Difficulty breathing and swallowing
- Inability to sit unsupported
- Type 2 (Intermediate Onset):
- Muscle weakness predominantly in the legs
- Ability to sit but not stand or walk independently
- Increased risk of respiratory infections
- Type 3 (Juvenile Onset):
- Mild to moderate muscle weakness
- Ability to stand and walk, but may lose these abilities over time
- Scoliosis and joint contractures
- Type 4 (Adult Onset):
- Mild muscle weakness and atrophy
- Slow progression of symptoms
- Typically remains ambulatory
Diagnosis
Diagnosis of SMA involves a combination of clinical evaluation, genetic testing, and electromyography (EMG):
- Clinical Examination: Assessment of muscle weakness, reflexes, and motor function.
- Genetic Testing: Identification of deletions or mutations in the SMN1 gene.
- Electromyography (EMG): Evaluation of the electrical activity of muscles to confirm the diagnosis.
Assistive Suggestions and Requirements
Requirement Percentage for Assistive Technology
Many patients with SMA will benefit from assistive technology, particularly those with significant motor impairments.
Assistive Technology Suggestions
- Mobility Aids:
- Wheelchairs: Manual or powered wheelchairs to support mobility and independence.
- Standing Frames: To promote weight-bearing and improve bone health.
- Communication Aids:
- Speech Generating Devices (SGDs): For individuals with speech difficulties.
- Voice Amplifiers: To assist with weak speech.
- Home Modifications:
- Grab Bars and Handrails: To enhance safety and prevent falls.
- Adjustable Beds: To improve comfort and ease of movement.
- Feeding Aids:
- Adaptive Utensils: For individuals with reduced hand strength.
- Feeding Tubes: For those with severe swallowing difficulties.
Access Modalities
- Switch Access: For individuals with severe motor impairments to control communication devices and computers.
- Voice-Controlled Systems: Beneficial for those who retain good vocal strength.
- Touchscreen Devices: Useful for those with adequate hand dexterity.
Care Management and Therapeutic Techniques
Aims
- To manage symptoms and maintain the highest possible level of independence and quality of life.
- To provide supportive care and address complications through a multidisciplinary approach.
- To offer education and support to patients and caregivers.
SLP Suggestions
- Assessment and Intervention:
- Regular Speech and Swallowing Assessments: To monitor changes and adjust therapy plans accordingly.
- Swallowing Techniques: Teaching techniques to ensure safe swallowing and reduce the risk of aspiration.
- Voice Therapy: Exercises to maintain voice strength and clarity.
- Augmentative and Alternative Communication (AAC):
- Introduction of AAC devices early in the disease progression to ensure familiarity and ease of use as the disease progresses.
Special Educator Suggestions
- Cognitive Rehabilitation:
- Activities designed to enhance executive function, memory, and attention.
- Use of memory aids, such as notebooks or electronic organizers.
- Behavioral Strategies:
- Structured routines to manage apathy and maintain engagement in activities.
- Positive reinforcement to encourage participation and effort in tasks.
Occupational Therapist Suggestions
- Daily Living Skills:
- Training in the use of adaptive equipment for self-care activities (e.g., dressing, grooming).
- Techniques to conserve energy and manage fatigue.
- Home and Environmental Modifications:
- Assessing and modifying the home environment to ensure safety and accessibility.
- Recommendations for ergonomic furniture and tools to support independence.
Recommendations on AAC and Other Details
Cognitive abilities in SMA generally remain intact, making literacy-based AAC highly effective for most individuals. Motor limitations often necessitate alternative access methods rather than symbol-based approaches.
- Text-Based AAC:
- Text-to-speech apps and devices for individuals who retain good literacy skills.
- Predictive text features to speed up communication.
- Symbol-Based AAC:
- Symbol-based systems like Picture Communication Symbols (PCS) for cognitive impairments affecting literacy.
- Dynamic display devices that adapt with the user's needs.
Additional Information
Spinal Muscular Atrophy is a genetic neuromuscular disorder that requires ongoing management and a multidisciplinary approach. Regular follow-ups with neurologists, pulmonologists, cardiologists, speech-language pathologists, occupational therapists, and special educators are essential. Patient and caregiver education, as well as access to community resources and support groups, can provide valuable assistance in managing this complex condition.

Extended Information
Pathological Insights and Disease Mechanism
Spinal Muscular Atrophy (SMA) is characterized by the degeneration of motor neurons in the spinal cord and brainstem, leading to progressive muscle weakness and atrophy. The survival motor neuron 1 (SMN1) gene is crucial for the production of the SMN protein, which is essential for the survival of motor neurons. The complete loss of SMN protein is embryonically lethal, whereas reduced levels result in the selective death of motor neurons, leading to muscle paralysis. The exact mechanisms by which SMN deficiency leads to motor neuron death are still under investigation, but it is known to affect RNA processing, axonal transport, and neuromuscular junction maintenance.
Genetic and Environmental Factors
SMA is caused by deletions or mutations in the SMN1 gene, which is inherited in an autosomal recessive pattern. This means that both copies of the gene must be affected for the disease to manifest. Carriers of a single mutated gene do not show symptoms but can pass the mutation to their offspring. Genetic testing can identify carriers and help with family planning. There are no known environmental factors that increase the risk of developing SMA.
Clinical Presentation and Disease Progression
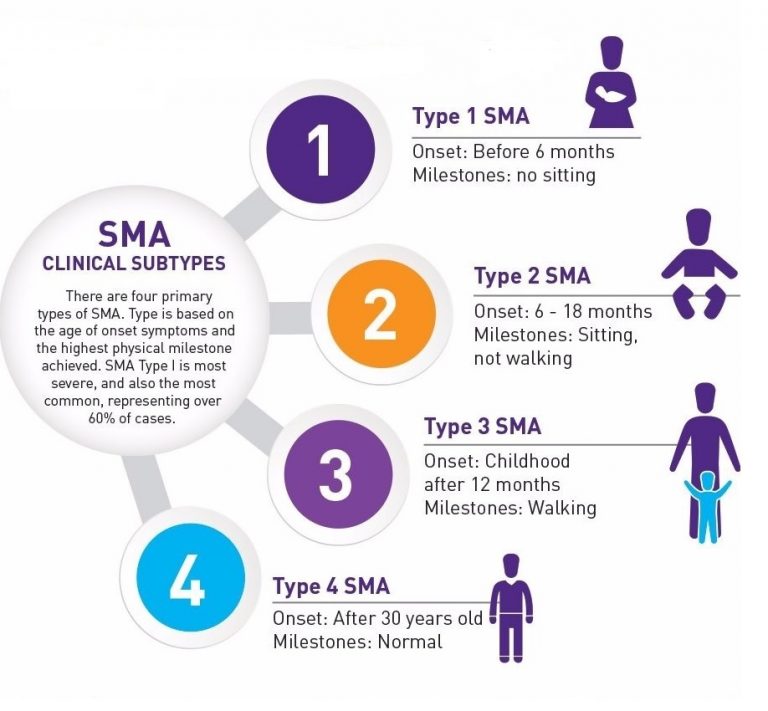
The clinical presentation of Spinal Muscular Atrophy (SMA) varies widely among individuals and is classified into four main types based on the age of onset and the highest motor function achieved:
- Type 1 (Infantile Onset): The most severe form, presenting within the first six months of life with severe muscle weakness, hypotonia (floppy baby syndrome), and difficulty breathing and swallowing. These infants are unable to sit unsupported and have a shortened life expectancy.
- Type 2 (Intermediate Onset): Presents between 6 and 18 months of age with muscle weakness predominantly in the legs. These children can sit but not stand or walk independently. They are at increased risk of respiratory infections and other complications.
- Type 3 (Juvenile Onset): Presents after 18 months of age with mild to moderate muscle weakness. These individuals can stand and walk but may lose these abilities over time. Scoliosis and joint contractures are common.
- Type 4 (Adult Onset): The mildest form, presenting in adulthood with mild muscle weakness and atrophy. Symptoms progress slowly, and individuals typically remain ambulatory.
Differential Diagnosis and Overlapping Syndromes
Diagnosing SMA involves differentiating it from other neuromuscular disorders that can present with similar symptoms. Conditions such as muscular dystrophies, congenital myopathies, and metabolic myopathies need to be considered. A thorough clinical evaluation, including a detailed history, physical examination, and appropriate diagnostic tests, is essential for an accurate diagnosis. Genetic testing for SMN1 gene mutations confirms the diagnosis and differentiates SMA from other conditions.
Therapeutic Interventions and Symptom Management
The management of SMA involves a combination of pharmacological and non-pharmacological treatments aimed at improving motor function, preventing complications, and enhancing the quality of life. Early intervention and a multidisciplinary approach are crucial for best outcomes.
Pharmacological Treatments:
- Nusinersen (Spinraza): An antisense oligonucleotide that increases the production of functional SMN protein by modifying the splicing of SMN2 pre-mRNA. It is administered intrathecally and has been shown to improve motor function and survival in individuals with SMA.
- Onasemnogene Abeparvovec (Zolgensma): A gene therapy that delivers a functional copy of the SMN1 gene using an adeno-associated virus vector. It is administered as a one-time intravenous infusion and is approved for use in children under two years of age.
- Risdiplam (Evrysdi): An orally administered small molecule that increases SMN protein levels by modifying SMN2 pre-mRNA splicing. It is approved for use in individuals with SMA aged two months and older.
Non-Pharmacological Treatments:
- Respiratory Support: Individuals with SMA, particularly those with Type 1 and Type 2, may require respiratory support to manage breathing difficulties. Non-invasive ventilation (e.g., BiPAP) and mechanical ventilation may be necessary to ensure adequate oxygenation and ventilation.
- Nutritional Support: Proper nutrition is essential for growth and overall health. Individuals with SMA may have difficulty swallowing and may require feeding tubes (e.g., gastrostomy tubes) to ensure adequate caloric intake and prevent aspiration.
- Physical Therapy: Physical therapy is crucial for maintaining muscle strength, flexibility, and joint mobility. Therapists work with patients to develop individualized exercise programs and recommend assistive devices to support mobility and independence.
- Occupational Therapy: Occupational therapy focuses on improving fine motor skills and enhancing the ability to perform daily activities. Therapists may recommend adaptive equipment and techniques to assist with self-care tasks.
Supportive Therapies:
- Speech and Language Therapy: Speech and language therapy is important for individuals with communication difficulties. Therapists work on improving speech clarity, language skills, and swallowing function. Augmentative and Alternative Communication (AAC) devices may be introduced to support communication for individuals with severe speech impairments.
- Orthopedic Interventions: Scoliosis and joint contractures are common in individuals with SMA. Orthopedic interventions, including bracing and surgical procedures, may be necessary to manage these complications and improve quality of life.
Lifestyle and Home Management:
- Regular Physical Activity: Maintaining a high level of fitness through regular physical activity is important for overall health and well-being. Exercise can help improve cardiovascular health, muscle strength, and mental health.
- Healthy Diet: A balanced diet rich in nutrients supports overall health and can help manage some of the symptoms associated with SMA. It is important to consult with a healthcare provider or nutritionist to develop an appropriate diet plan.
Patient and Caregiver Education:
- Education and Training: Providing education and training to patients and caregivers is crucial for effective management of SMA. This includes information on the condition, treatment options, symptom management, and the use of assistive devices.
- Support Groups: Joining support groups can provide valuable emotional support and practical advice from others who are experiencing similar challenges. These groups can also offer a sense of community and reduce feelings of isolation.
Research and Future Directions:
Research into Spinal Muscular Atrophy is ongoing, with a focus on understanding the underlying mechanisms of the disease and developing new treatments. Advances in genetic research, neuroimaging, and therapeutic interventions hold promise for improving the management and outcomes of SMA. Clinical trials and research studies are essential for advancing knowledge and finding more effective treatments.
In conclusion, Spinal Muscular Atrophy is a complex genetic disorder that requires a comprehensive, multidisciplinary approach to management. Early diagnosis, individualized treatment plans, and ongoing support are essential for improving the quality of life for individuals with SMA. By staying informed about the latest research and treatment options, patients and caregivers can work with healthcare providers to achieve the best possible outcomes.
References
- , et al. (2016). Commonality amid diversity: Multi-study proteomic identification of conserved disease mechanisms in spinal muscular atrophy. Neuromuscular Disorders. https://doi.org/10.1016/j.nmd.2016.06.004
- , & . (2015). Spinal Muscular Atrophy Therapeutics: Where do we Stand?. Neurotherapeutics. https://doi.org/10.1007/s13311-015-0337-y
- , & . (2014). The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics. https://doi.org/10.1007/s13311-014-0314-x
- , et al. (2014). Molecular mechanisms and animal models of spinal muscular atrophy. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. https://doi.org/10.1016/j.bbadis.2014.07.024
- , et al. (2011). Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. European Journal of Human Genetics. https://doi.org/10.1038/ejhg.2011.134
- . (2008). Carrier screening for spinal muscular atrophy. Genetics in Medicine. https://doi.org/10.1097/GIM.0b013e318188d069
- jedrzejowska. (2009).
- bach. (2000).
Epidemiology and Demographics
Etiology and Pathophysiology
What causes Spinal Muscular Atrophy?
What does Spinal Muscular Atrophy do to the body?
Clinical Features and Stages
Diagnosis
Diagnostic Criteria
Genetic Testing
Differential Diagnosis
Assistive Technology and AAC Interventions
Communication Devices
Mobility Aids
Access Modalities
Environmental Control Units
Clinical Recommendations
For Speech-Language Pathologists
For Occupational Therapists
For Physical Therapists
For Applied Behavior Analysts
For Special Educators
For All Staff and Caregivers
Care Management
Medical Management
Positioning and Handling
Feeding and Swallowing
Psychosocial Support
Educational Support
IEP Goal Examples
Accommodations and Modifications
Transition Planning
Support and Resources
🏛️ Foundations and Research
🌐 Online Communities
📚 Educational Resources
💰 Financial Assistance
References
Disclaimer: This comprehensive clinical guide is designed for healthcare professionals, educators, and families. For specific medical advice, please consult with qualified healthcare providers.